
Pertinent Parameters Selection for Processing of Short
Amino Acid Sequences
Zbigniew Szymański
1
, Stanisław Jankowski
2
, Marek Dwulit
1
Joanna Chodzyńska
3
and Lucjan S. Wyrwicz
3
1
Warsaw University of Technology, Institute of Computer Science
ul. Nowowiejska 15/19, 00-665 Warszawa, Poland
2
Warsaw University of Technology, Institute of Electronic Systems
ul. Nowowiejska 15/19, 00-665 Warszawa, Poland
3
Maria Skłodowska-Curie Memorial Cancer Center and Institute of Oncology
Laboratory of Bioinformatics and Systems Biology, ul. Roentgena 5
02-781 Warszawa, Poland
Abstract. The paper describes the Least Squares Support Vector Machine (LS-
SVM) classifier of short amino acid sequences for the recognition of kinase-
specific phosphorylation sites. The sequences are represented by the strings of
17 characters, each character denotes one amino acid. The data contains se-
quences reacting with 6 enzymes: PKA, PKB, PKC, CDK, CK2 and MAPK.
To enable classification of such data by the LS-SVM classifier it is necessary to
map symbolic data into real numbers domain and to perform pertinent feature
selection. Presented method utilizes the AAindex (amino acid index) set up of
values representing various physicochemical and biological properties of amino
acids. Each symbol of the sequence is substituted by 193 values. Thereafter the
feature selection procedure is applied, which uses correlation ranking formula
and the Gram-Schmidt orthogonalization. The selection of 3-17 most pertinent
features out of 3281 enabled successful classification by the LS-SVM.
1 Introduction
The paper presents the method of recognition of kinase-specific phosphorylation sites
by the Least Squares Support Vector Machine (LS-SVM) classifier of short amino
acid sequences [1]. Protein phosphorylation as a chemical modification of amino acid
side chains plays a significant role in cell signaling. Phosphorylation is performed by
an addition of a phosphate (PO
4
) group to specific substrate sites performed by spe-
cific enzymes known as protein kinases. This post-translational
modification of pro-
teins is essential for correct functioning of every cellular process including metabol-
ism, growth and differentation. Phosphorylation can affect activity of enzymes and
defects in protein kinase function may lead to various diseases including cancer.
Szymanski Z., Jankowski S., Dwulit M., ChodzyÅ
ˇ
Dska J. and S. Wyrwicz L.
Pertinent Parameters Selection for Processing of Short Amino Acid Sequences.
DOI: 10.5220/0003040600250032
In Proceedings of the 10th International Workshop on Pattern Recognition in Information Systems (ICEIS 2010), page
ISBN: 978-989-8425-14-0
Copyright
c
2010 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
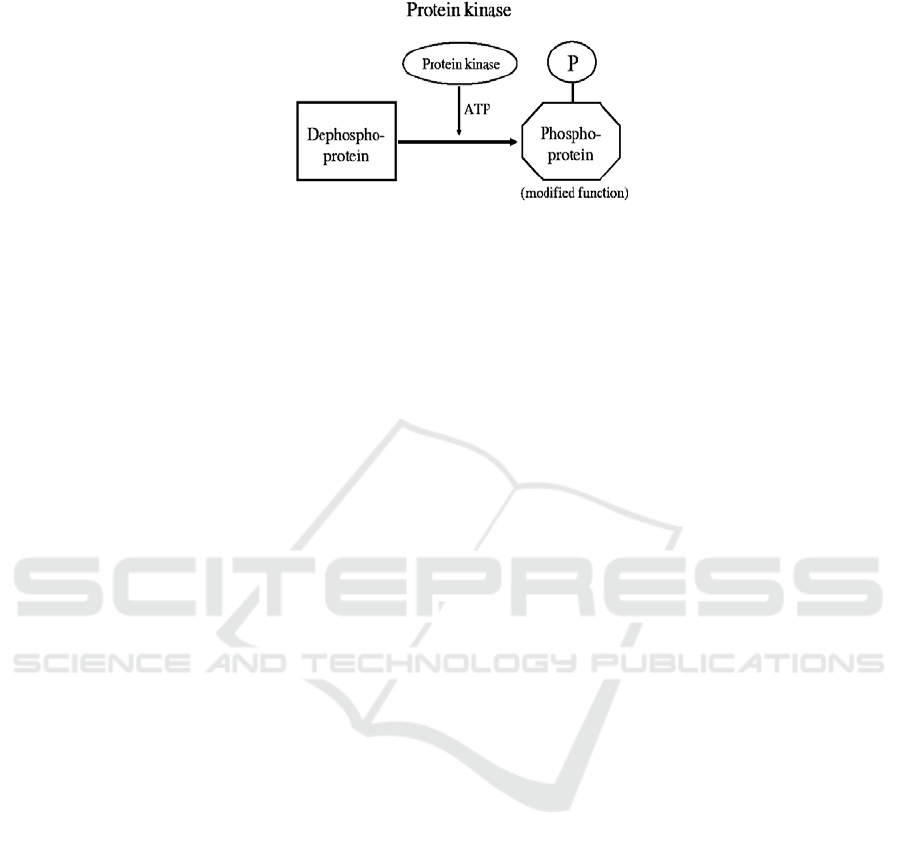
Fig. 1. Chemical mechanism of phosphorylation.
It is estimated that nearly 30% of human proteome is phosphorylated at any time
by more than 500 protein kinases encoded by the genome [2]. Within a particular
protein an event of phosphorylation can occur on multiple various sites, occurring
mainly on side chains of serine (S), threonine (T) or tyrosine (Y). Still we have li-
mited biochemical understanding of the process of protein phosphorylation and expe-
rimental verification of given phosphorylation site is very difficult and time consum-
ing. Therefore this problem cannot easily be addressed to classification algorithms,
since we cannot confirm a negative dataset (i.e. amino acids of given type which nev-
er undergo phosphorylation). As mentioned above – with more than 500 different
protein kinases in human cells, each possessing a different profile of activities against
biological target, there is a need to develop a methods for better understanding of
kinase biology. Also general rules governing specificities of protein kinases remain
unknown. Therefore many in silico methods for identifying protein phosphorylation
sites have been proposed.
Existing approaches differ in classification methods, training sets as well as types
of results. The KinasePhos web server applies a hidden Markov model for learning of
sequences surrounding to the phosphorylation residues to predict phosphorylation
sites and related kinases [3]. NetPhos uses a neural network based on sequences of
protein substrates and information about local tertiary structure near the phosphoryla-
tion sites [4]. Scansite 2.0 is a web tool developed by Yaffe et al. [5]. It compares a
given sequence to short protein motifs obtained from peptide libraries and represented
as position-specific scoring matrices.
The presented approach deals with a problem of recognition of various substrates
by specified kinases in order to create a “cross-classifier” by testing peptides known
to be modified by a given kinase (positives) versus other peptides phosphorylated by
other kinases. Phosphorylation sites categorized by corresponding annotated protein
kinases were derived from the Phospho.ELM database [6]. The amino acid sequences
are represented by the strings of 17 characters, each character denotes one amino ac-
id. To enable classification of such data by the LS-SVM classifier it is necessary to
map symbolic strings into real numbers domain. Statistical classifiers (e.g. LS-SVM)
have to meet basic mathematical requirements. It can be concluded from the T. Cover
theorem [7] that the number of elements N of the learning data set has to be greater
than 2(d+1), where d is the number of features. If the learning data set does not satis-
fy this theorem, the obtained generalization of the classifier is equivalent to randomly
defined classifier. Therefore it is necessary to perform feature selection and to restrict
the whole data set only to the subset based on most relevant variables.
26

Several methods have been developed for mapping of symbolic amino acid se-
quences to real numbers domain. There are very simple methods like binary encoding
[8] producing very large feature vectors, where no biochemical knowledge is utilized.
More advanced methods exploit biochemical knowledge e.g. the Blosum 62 substitu-
tion matrix [9]. However the size of feature vector may be still large like in Blosum
representation [8]. In such cases a large training data set is required to create a statis-
tical classifier.
The presented method utilizes the AA index (amino acid index) [10] set up of val-
ues representing various physicochemical and biological properties of amino acids.
Each symbol from the amino acid sequence is substituted by the corresponding values
from the AAindex. Thereafter the feature selection procedure is applied, which uses
simple ranking formula and the Gram-Schmidt orthogonalization [11,12]. Next, the
obtained data set is used as input to the LS-SVM classifier.
The method described in this paper is aimed toward the research of enzymes struc-
ture. The identification of types and positions of amino acids sequences that define
the ability of reaction with selected enzymes can be useful for building of three di-
mensional enzyme models. The long term goal of our research is the design of a clas-
sifier able to predict if an amino acid sequence can react with a given enzyme.
2 Input Data
The data set contains the 17-symbols amino acids sequences grouped with respect to
their reactions with 6 selected enzymes. The data set was derived at the Maria
Skłodowska-Curie Memorial Cancer Center and Institute of Oncology in Warsaw.
The data file is in the text form. An example of the input file format is shown in Fig.
2. A line starting with the “#” sign denotes the line describing an enzyme symbol.
A line starting with a letter contains a sequence of 17 amino acids.
A line of enzyme symbol opens a new series of amino acids sequences reacting
with this particular enzyme. E.g. the sequence SKSSPKDPSQRRRSLEP reacts with
#PKC
SKSSPKDPSQRRRSLEP
RRSRRYRRSTVARWRRR
RRRRSRRSTVAWRRRRV
#CK2
RRRRSRRVSRRRRARRR
RRRRPRSVSRRWRARRR
RRSRRYRRSTVARWRRR
RTSAVPTLSTFRTTRVT
Fig. 2. Example data from the input file. Lines starting with ‘#’ denote class names.
PKC enzyme. Respectively, the amino acids sequence RRRSRRVSRRRRARRR
reacts with CK2 enzyme.
The data file contains short amino acid sequences reacting with 6 enzymes: PKA,
PKB, PKC, CDK, CK2 and MAPK. Our data set comprised 1641 data samples. The
27

number of samples belonging to 6 analyzed classes: PKA – 322, PKB – 83, PKC –
382, CDK – 325, CK2 – 280, MAPK- 249.
The goal of this project is to obtain the statistical classifier that is able to divide the
amino acids sequences into 6 classes. It is important to notice that one sequence can
belong to more than one class. For example the sequence
RRSRRYRRSTVARWRRR belongs to either PKC or CK2 class, as this sequence
reacts with both enzymes.
3 Method
The proposed approach consists of two stages. Mapping of amino acid symbols into
real numbers is performed in the first stage. Each symbol is substituted by corres-
ponding values from the AAindex data set. In order to decrease number of features
only 193 uncorrelated indices were chosen for the substitution, out of 544 indices.
Then each amino acid sequence is described by 3281 (17x193) features – most of
them are irrelevant for classification purposes.
It is clear that the learning data set of 984 data samples (and 3281 variables) does
not meet requirements of the Cover theorem. The goal of the second stage is selection
of relevant features (variables). The ranking by correlation and the Gram-Schmidt
orthogonalization is used to solve the task. Results of the second stage are applied to
the statistical classifiers - the least-squares support vector machine (LS-SVM) [1, 11].
3.1 AAindex based Mapping of Symbols
An amino acid index [10, 13] is a set of 20 numerical values representing various
physicochemical and biological properties of amino acids. The AAindex1 section of
the Amino Acid Index Database is a collection of published indices together with the
result of cluster analysis using the correlation coefficient as the distance between two
indices.
H ARGP820102
D Signal sequence helical potential (Argos et al., 1982)
R LIT:0901079b PMID:7151796
A Argos, P., Rao, J.K.M. and Hargrave, P.A.
T Structural prediction of membrane-bound proteins
J Eur. J. Biochem. 128, 565-575 (1982)
C ARGP820103 0.961 KYTJ820101 0.803 JURD980101
0.802
I A/L R/K N/M D/F C/P Q/S E/T G/W H/Y I/V
1.18 0.20 0.23 0.05 1.89 0.72 0.11 0.49 0.31 1.45
3.23 0.06 2.67 1.96 0.76 0.97 0.84 0.77 0.39 1.08
Fig. 3. Example entry from the AAindex1 data set.
The meaning of the fields in an AAindex1 entry [13]: H - accession number; D -
data description; R - LITDB entry number; A - author(s); T - title of the article; J -
28

journal reference; C - accession numbers of similar entries with the correlation coeffi-
cients of 0.8 (-0.8) or more (less); I - amino acid index data in the following order:
Ala Arg Asn Asp Cys Gln Glu Gly His Ile
Leu Lys Met Phe Pro Ser Thr Trp Tyr Val
3.2 Features Ranking Method
The orthogonalization procedures enable us the ranking of the influence of every in-
put feature on the class label. The presented method uses the ranking by correlation
coefficient and the Gram-Schmidt orthogonalization procedure for pointing out the
most salient features of classifier [11,12].
The set of N input-output pairs (measurements of the output of the phenomenon to
be modeled, and of the candidate features) is available. We denote by: Q – number of
candidate features; N – number of measurements of the process to be modeled;
x
i
=[x
i
1
, x
i
2
, ...x
i
N
,] – the vector of the i-th feature values of N measurements; y
p
– the
N-dimensional vector of the class labels.
We consider the NxQ matrix X=[x
1
, x
2
, ..., x
Q
,]. The ranking procedure starts with
calculating the square of correlation coefficient:
cos
2
(x
k
,y
p
)=<x
k
y
p
>
2
/(||x
k
||
2
||y
p
||
2
)
(1)
The greater it is, the better the k-th feature vector explains the y
p
variation.. As the
first basis vector we indicate the one with the largest value of correlation coefficient.
All the remaining candidate features and the output vector are projected onto the null
subspace (of dimension N-1) of the selected feature. Next, we calculate correlation
coefficients for the projected vectors and again indicate the one with the largest value
of this quantity. The remaining feature vectors are projected onto the null subspace of
the first two ranked vectors by the classical Gram-Schmidt orthogonalization. This
procedure is continued until all the vectors x
k
are ranked.
To reject the irrelevant inputs we compare its correlation coefficient with that of a
random probe. The remaining features are considered relevant to the model.
3.3 LS-SVM Classifier
LS-SVM originates by changing the inequality constraints in the SVM formulation to
equality constraints with objective function in the least squares sense [1]. Data set D
is defined as:
)},{(
ii
tD x=
}1,1{, +−∈⊂∈
i
d
i
tRXx
(2)
The LS-SVM classifier performs the function:
bf
+
=
)()( xwx
φ
(3)
This function is obtained by solving the following optimization problem:
29

∑
=
−−+=
l
i
ii
bt
1
22
])([||||
2
1
xww
φγ
L
(4)
Hence, the solution can be expressed as the linear combination of kernels weighted
by the Lagrange multipliers
α
i
:
∑
=
+=
l
i
ii
bKf
1
),()( xxx
α
(5)
The global minimizer is obtained in LS-SVM by solving the set of linear equations
⎥
⎦
⎤
⎢
⎣
⎡
=
⎥
⎦
⎤
⎢
⎣
⎡
⎥
⎥
⎦
⎤
⎢
⎢
⎣
⎡
+
−
0
0
!
tα
1
1IK
b
T
γ
(6)
In this work the RBF kernel is applied:
γη
/1},||'||exp{)',(
2
=−−= σK xxxx
(7)
The parameters σ and γ are adjusted upon the class and the number of input va-
riables. This system is easier to solve as compared to SVM. However the sparseness
of the support vectors is lost. In SVM, most of the Lagrangian multipliers α
i
are equal
0 while in LS-SVM the Lagrangian multipliers α
i
are proportional to the errors e
i.
4 Results
The tests were performed on 20 data sets randomly generated from the data set con-
taining all sequences. The 60% of data samples were used for the training of the clas-
sifier. Remaining data samples were used for validation of obtained LS-SVM model.
For each enzyme the binary classification was performed - one against all by a sepa-
rate classifier.
Table 1. Number of features used for classification.
Class Name
No of Relevant
Positions
No of Features
PKA
2 16
PKB
1 5
PKC
2 12
CDK
1 3
CK2
2 17
MAPK
1 17
The number of relevant features (Table 1) calculated by the model variables
ranking procedure varies from 3 (CDK class) to 17 (CK2, MAPK classes). The se-
30
lected relevant features correspond to 1 or 2 relevant positions in the original amino
acid sequence. Table 2 contains the results of the model variables ranking procedure
for the CDK class. Three features contributing to the recognition of CDK class cor-
respond to 10
th
position in the amino acid sequence.
Table 2. Features selected for classification of CDK class.
Feature No. Sequence Position
AAindex1 Accession
Number
1 10 ARGP820102
2 10 CHAM830104
3 10 QIAN880116
The summary of performed research is presented in Table 3. The classifier perfor-
mance for MAPK class is lower than for the other classes. This fact may be caused by
the mapping procedure. After substitution different amino acid sequences may be
represented by the same feature vector. This is one of major drawbacks of the me-
thod. The balance between precision and recall may be slightly modified by different
selection of hyperparameters of the LS-SVM classifier and number of variables.
Table 3. Classification results.
Class name
Precision ±
σ[%]
Recall ± σ[%] Total accuracy ± σ [%]
PKA
64,03 ± 5,06 48,41 ± 5,26 84,53 ± 1,10
PKB
33,97 ± 6,51 83,66 ± 5,19 89,81 ± 2,37
PKC
67,27 ± 4,74 60,71 ± 3,29 83,98 ± 1,15
CDK
54,32 ± 1,69 95,61 ± 1,11 83,07 ± 0,93
CK2
75,01 ± 4,48 59,93 ± 4,42 89,72 ± 1,02
MAPK
26,21 ± 12,26 71,39 ± 37,00 71,51 ± 5,10
The standard deviations calculated for precision and recall of the MAPK class
stand out from the values calculated for other classes. It may be caused by the non
uniform nature of this class, which could be divided into separate subclasses.
5 Conclusions
The presented feature selection method reduces the number of considered features
from 3281 to an acceptable amount: 3-17 features. At the preprocessing stage 351
mutually correlated features from the AAindex1 data set were removed. Hence, the
obtained statistical classifiers satisfied the Cover theorem. The mutually correlated
features were removed by the procedure based on the Gram-Schmidt orthogonaliza-
tion. The relevant features correlated with target labels were included into the ana-
lyzed data set. It can be concluded that the presented pertinent feature selection me-
31

thod increased the probability of successful classification. The presented method will
be applied in the research of enzymes structure. The identification of amino acids
chemical properties with respect to selected enzymes can be useful for building three
dimensional molecular models.
References
1 Suykens J.A.K and Vandewalle J.: Least squares support vector machine classifier, Neural
Processing Letters, 9 (1999), 293-300
2 Wan J., Kang S., Tang C., Yan J., Ren Y., Liu J., Gao X., Banerjee A., Ellis L.B.M., and Li
T.: Meta-prediction of phosphorylation sites with weighted voting and restricted grid
search parameter selection, Nucleic Acids Res. 36(4): e22, 2008
3 Hsien-Da H., Tzong-Yi L., Shih-Wei T. Jorng-Tzong H.: KinasePhos: a web tool for iden-
tifying protein kinase-specific phosphorylation sites, Nucleic Acids Res. 33(Web Server Is-
sue), W226-W229, 2005
4 Blom N., Gammeltoft S., Brunak S.: Sequence and structure-based prediction of eukaryotic
protein phosphorylation sites, J. Mol. Biol. 294, 1351-1362, 1999
5 Obenauer J.C., Cantley L.C., Yaffe M.B.: Scansite 2.0: Proteome-wide prediction of cell
signaling interactions using short sequence motifs, Nucleic Acids Res. 31, 3635-3641, 2003
6 Phospho.ELM database, http://phospho.elm.eu.org/
7 Cover T.M.: Geometrical and statistical properties of systems of linear inequalities with
applications in pattern recognition, IEEE Trans. on Electr. Comp., 1965 EC14, 326-334
8 Plewczynski D., Tkacz A., Wyrwicz L.S., Godzik A., Kloczkowski A., Rychlewski L.:
Support-vector-machine classification of linear functional motifs in proteins, J Mol Model
(2006) 12: 453–461
9 Henikoff S., Henikoff J.G.: Amino acid substitution matrices from protein blocks. Proc.
Natl. Acad. Sci. USA, Vol. 89, pp. 10915-10919, November 1992
10 Kawashima S. and Kanehisa M.; AAindex: amino acid index database. NucleicAcids Res.
28, 374 (2000)
11 Stoppiglia H., G. Dreyfus, R. Dubois, Y. Oussar, Ranking a Random Feature for Variable
and Feature Selection, Journal of Machine Learning Research 3 (2003), 1399-1414
12 Jankowski S., Szymański Z., Raczyk M., Piatkowska-Janko E., Oreziak A. Pertinent sig-
nal-averaged ECG parameters selection for recognition of sustained ventrical tachycardia.
XXXVth International Congress on Electrocardiology, 18-21 September, 2008, St. Peters-
burg, Russia, pp. 43 (abstract).
13 Kawashima S., AAindex: Amino Acid Index Database Release 9.1, Aug 2006,
ftp://ftp.genome.jp/pub/db/community/aaindex/aaindex.doc
32
