
EFFICIENT PATH KERNELS FOR REACTION FUNCTION
PREDICTION
Markus Heinonen
∗
, Niko V
¨
alim
¨
aki
∗
, Veli M
¨
akinen and Juho Rousu
Department of Computer Science, University of Helsinki, Helsinki, Finland
Keywords:
Graph kernels, Compressed data structures, XBW transform, Reaction graph, Hierarchical classification.
Abstract:
Kernels for structured data are rapidly becoming an essential part of the machine learning toolbox. Graph
kernels provide similarity measures for complex relational objects, such as molecules and enzymes. Graph
kernels based on walks are popular due their fast computation but their predictive performance is often not
satisfactory, while kernels based on subgraphs suffer from high computational cost and are limited to small
substructures. Kernels based on paths offer a promising middle ground between these two extremes. However,
the computation of path kernels has so far been assumed computationally too challenging. In this paper we
introduce an effective method for computing path based kernels; we employ a Burrows-Wheeler transform
based compressed path index for fast and space-efficient enumeration of paths. Unlike many kernel algorithms
the index representation retains fast access to individual features. In our experiments with chemical reaction
graphs, path based kernels surpass state-of-the-art graph kernels in prediction accuracy.
1 INTRODUCTION
Kernel methods have proven an efficient approach in
machine learning tasks, such as classification, regres-
sion and clustering (Shawe-Taylor and Christianini,
2004). A kernel defines a similarity function between
two objects, which corresponds to implicitly mapping
the objects to a feature space, in which a dot product
equals the kernel value. This property of kernels is
especially suitable for complex structural data, as the
object’s vectorial representation is formed automati-
cally.
Graph kernels try to capture the essential topo-
logical features of the graph by providing a (possi-
bly infinite) feature vector representation of the graph,
and subsequently computing the similarity between
the feature vectors. Paired with kernel methods, the
relevant application-dependent substructure signals
can be exploited. Graph kernels can be categorised
based on the generality of the feature class: sequence
based kernels enumerate e.g. walks or paths, subtree
based kernels allow sequences to branch, and sub-
graph based kernels place no restrictions on the sub-
structure.
The commonly used walk kernels can be com-
puted efficiently up to infinite length, but they contain
∗
Equal contribution from these authors
arbitrary repeats of themselves (G
¨
artner, 2003). Any
walk can continue infinitely by traversing itself back
and forth. These repeated walks contain no additional
information about the graph, however they result in
infinite feature space and in kernels with large norms.
Down weighting longer walks alleviates the problem
(G
¨
artner, 2003). In (Mahe et al., 2005) the problem
was fought by introducing non-tottering walks, where
tottering cycles of length two are prohibited in walks.
Kernels based on paths, that is walks with no rep-
etitions, are more challenging to compute and few
practical algorithms so far exists. In (Borgwardt et al.,
2005) an efficient kernel using shortest paths between
nodes in the graphs was proposed. The shortest paths
kernel has an exceptionally small feature space as
there only exists n
2
shortest paths in a graph of size
n, which makes its efficient to compute. In (Ralaivola
et al., 2005) a suffix trees was utilized in computing
the pairwise path kernels individually up to length 10.
However, so far a method for efficiently computing
an all-paths kernel has been missing. In this paper we
propose an efficient method for computing path ker-
nels using a compressed path index as an intermediate
data structure. The compressed path index is based on
a Burrows-Wheeler transform of labeled trees (Fer-
ragina et al., 2009) and contains the feature values ex-
plicitly which allows for various kernel functions in
the feature space.
202
Heinonen M., Välimäki N., Mäkinen V. and Rousu J..
EFFICIENT PATH KERNELS FOR REACTION FUNCTION PREDICTION.
DOI: 10.5220/0003779402020207
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2012), pages 202-207
ISBN: 978-989-8425-90-4
Copyright
c
2012 SCITEPRESS (Science and Technology Publications, Lda.)

We take two steps to compute path kernels when
given a set of graphs as an input: First, we create a
specialized path index that represents all the paths,
up to some maximum length, of all graphs in com-
pressed form and allows us to efficiently traverse all
unique paths and output their frequencies for desired
graphs. Then, the path frequencies are used to com-
pute path kernels via inner products. In our experi-
ments we compare path based graph kernels against
walk based kernels, shortest paths based kernels, as
well as the custom reaction graph kernel by (Saigo
et al., 2010). The performance is evaluated in a hi-
erachical multilabel classification task of assigning a
biochemical reaction to the correct branch of the En-
zyme Commision (EC) hierarchy.
In section 2 we review existing sequence based
kernels on graphs, including the shortest paths kernel.
In section 3 the compressed path index for the com-
putation of path kernels is introduced. In section 4
experiments are carried out where the path based ker-
nels are compared against state-of-the-art graph ker-
nels. We also review the run time performance of our
method. We conclude with discussions in section 5.
2 GRAPH KERNELS
In this section we review existing approaches for com-
puting graph kernels. We consider a labeled undi-
rected graph G = (V, E,L) with n nodes, where a la-
beling function L applies to both nodes v ∈ V and
edges (v,u) ∈ E. A walk w is a sequence of adjacent
vertices. A path p is a walk where no node repeats.
Random walk kernels compute the weighted sum
of matching walks in a pair of graphs, utilizing either
the adjacency matrix exponential (G
¨
artner, 2003) or
a markov process model (Kashima et al., 2003). If
the contribution of each walk is downscaled appropri-
ately according to its length k with λ
k
the walk kernel
can be computed until convergence in cubic time. An
generalization to marginalized kernels prevents non-
tottering walks by utilizing second order markov pro-
cesses (Mahe et al., 2005).
With λ < 1 the contribution of longer walks
quickly becomes negligible and long walks have ef-
fectively no effect on the kernel value in the stan-
dard walk kernel. This is sometimes against what
we desire – longer walks may contain important in-
formation for e.g. graphs with repetitive substruc-
tures, where the walk length is required to surpass
the diameter of the substructure to notice the repeti-
tion. Therefore we consider finite-length walk ker-
nels, where walks up to length k are constructed ex-
plicitly. Working with explicit walks allows us to re-
gard paths and non-tottering walks. We can count the
number of matching k-length walks in two graphs by
using dynamic programming (Demco, 2009).
Borgwardt et al. defined a family of kernels
using special subsets of paths, namely the set
of shortest paths between all nodes (Borgwardt
et al., 2005). All pair shortest paths can be
computed with Floyd-Warshall algorithm in O(n
3
)
time. The kernel is then defined K
sp
(G,G
0
) =
∑
p∈S P (G),p
0
∈S P (G
0
)
K
l
(p, p
0
), where S P (G) is the set
of shortest paths between all nodes of graph G and K
l
is a positive semi-definite path matching kernel. How-
ever, in many applications it is not clear that shortest
paths carry a special meaning. Thus efficient methods
to compute path kernels with a broader feature space
is still desirable.
3 COMPUTING PATH KERNELS
VIA COMPRESSED PATH
INDEX
We wish to use the set of all paths to characterize a
given graph. We define an all paths kernel as a dot
product between shared path counts. Computing the
path kernel is known to be NP-hard (Borgwardt et al.,
2005). However, we will show here that it is possible
to compute them with typical molecular datasets such
as KEGG reaction database.
We use a four-fold procedure to compute the ker-
nel. First, trees up to height h of all graphs are enu-
merated using each node subsequently as the root
node. Then, the resulting trees are used as an input to
a path index construction. Finally, the feature values
are extracted from the index, and the kernel is explic-
itly computed.
Path Enumeration. For each vertex u ∈ V , we
build a tree T
u
that contains all paths originating from
u. This can be done with a depth-first traversal in G,
and the result is a set of trees T = {T
u
| u ∈ V }. Fig-
ure 1 gives an example of a graph and one of the trees
generated. When multiple graphs G
1
,G
2
,.. .,G
g
are
given, each graph can be processed separately cre-
ating a set of tree sets T
1
,T
2
,.. .,T
g
. Path enumera-
tion can be generalized to directed and edge-labeled
graphs.
Path Index Construction. We store the set of trees
as a path-sorted transform dubbed XBWT (Ferragina
et al., 2009). We will show how the resulting index
can be used to efficiently compute path frequencies,
EFFICIENT PATH KERNELS FOR REACTION FUNCTION PREDICTION
203

A B
B
C
(a)
A
B
BB
c
B
C
b
b c
(b)
S
last
S
α
S
π
0 A empty
0 B A
0 B A
1 C A
1 B BA
0 b BA
1 c BA
1 c BBA
1 b BCA
1 B CA
(c)
Figure 1: (a) Example graph. (b) Paths originating from
node A. (c) An XBWT representation of the tree in (b).
The rows are lexicographically sorted by upward paths, S
π
,
which are displayed here for reference and not stored at any
point of the algorithm.
but first, let us describe how to construct the transform
for a single tree T of t nodes.
The XBWT of T is constructed in two phases
(Ferragina et al., 2009). The first phase starts by ini-
tializing an array S[1,t] to contain the nodes in pre-
order: for each node u, we store its label α[u], a parent
pointer and a boolean value last[u]. We set last[u] = 1
if u is the right-most child of its parent. Finally, we
need to be able to distinguish between internal and
leaf node labels — for example, in Figure 1, we use
lower-case labels for leaves. In the second phase, S
is stably sorted according to the lexicographical order
of node’s upward path π[u], that is, the labeled path
from u to the root. In (Ferragina et al., 2009) an op-
timal O(t)-time algorithm was given to sort S in im-
plicit manner, without a need to store the π[u] values
explicitly.
The final XBWT of T consists of a bitvector S
last
and an array of node labels, denoted by S
α
. They are
populated simply by following the order the nodes ap-
pear in S after the sorting phase. Figure 1(c) gives an
example of values of S
α
and S
last
for the tree in Fig-
ure 1(b). The actual index, which grants us naviga-
tional operations on T and efficient way of counting
path frequencies, requires rank and select queries on
S
α
and S
last
. The query rank
c
(S
α
,i) gives the number
of times c appears in S
α
[1,i], and select
c
(S
α
,i) gives
the position of the i-th c in S
α
. Both of these queries
can be solved in O(logσ/log logt) time, for any al-
phabet size σ = O(t
ε
) and ε < 1, by using a Wavelet
tree data structure requiring tH
0
(S
α
) + o(t log σ) bits
of memory (Ferragina et al., 2007), where H
0
(S
α
) ≤
logσ is the 0-th order entropy of S
α
Thus, the whole
index requires in total tH
0
(S
α
)+t + o(t) bits, plus an-
other t bits if we use an additional bitvector to dis-
tinguish leaf node labels. The XBWT representation
Algorithm traverse(s,e,P,l f ):
1 C ← Set of internal node symbols on S
α
[s,e].
2 L ← Set of leaf node symbols appearing on S
α
[s,e].
3 for each c ∈ C do
4 l f
0
← 0
5 if leaf(c) ∈ L then
6 L ← L \ leaf(c)
7 l f
0
← Number of leaf(c) in S
α
[s,e].
8 z
1
← select
c
(S
α
,rank
c
(S
α
,s − 1) + 1)
9 z
2
← select
c
(S
α
,rank
c
(S
α
,e))
10 s
0
← GetRankedChild(z
1
,1)
11 e
0
← GetRankedChild(z
2
,GetDegree(z
2
))
12 traverse(s
0
,e
0
,cP, l f
0
)
13 for each c ∈ L do
14 l f
0
← Number of leaf symbols c in S
α
[s,e].
15 Output subpath cP with frequency l f
0
.
16 f ← rank
1
(S
last
,e) − rank
1
(S
last
,s − 1)
17 Output subpath P with frequency f + l f .
Figure 2: Recursive algorithm to traverse through all
uniquely labeled, root-originating paths. The first call of
the recursion is traverse(1, 1, empty,0). Function leaf(c)
is just a simple conversion between internal and leaf node
labels, e.g. an upper-case conversion in Figure 1.
generalizes to multiple trees quite naturally — we
omit the technical details here for brevity.
In (Ferragina et al., 2009) a comprehensive list of
navigational operations supported by the index were
introduced. Due to the lexicographical ordering of
S, all children of an internal node occur at a specific
range [s,e] in S. For example, children of the root
node in Figure 1(c) can be found on rows [2,4]. Oper-
ation GetChild(i) returns the range [s,e] correspond-
ing to all the children of S[i] in O(log σ/ log log n)
time (we omit details here for brevity) (Ferragina
et al., 2009). Similarly, GetRankedChild(i, j) returns
the j-th child of i, which is simply s + j − 1. The de-
gree of node i is GetDegree(i) = e − s +1 where [s,e]
is given by GetChild(i).
Computing Path Frequencies. Next we will give
a recursive algorithm to output, for all unique root-
originating paths, the number of times the path oc-
curs, i.e. path’s frequency, in each tree. For now, as-
sume that we are given an index containing one tree T .
At the start of the recursion, we initialize the current
subpath P to be empty, and set s = e = 1. The follow-
ing invariant holds throughout the recursion: at every
step, the range S[s,e] corresponds to all nodes having
upward path equal to P.
Each recursion step proceeds as follows. First we
enumerate all unique labels appearing on the range
S
α
[s,e] by using a range search on the Wavelet tree
(cf. rows 1–2 in Figure 2). The labels can branch ei-
ther to internal nodes or leaf nodes, or both. For each
label c that branches with internal nodes, we locate
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
204

the first and last occurrence of c in S
α
[s,e] (rows 8–
9) and take their first and last child as the new range
(rows 10-11). Then a recursive call is done for the
new range [s
0
,e
0
] and subpath P
0
= cP. However, if
there are also leaf nodes branching with c, their fre-
quencies must be passed on for the recursive call since
leaves that match the path cP appear on S[s,e] in-
stead of the new interval (rows 5–7). Furthermore,
remaining leaf symbols must be handled separately
(rows 13–15). Finally, we output the frequency of the
current subpath P, which equals the number of 1-bits
in S
last
[s,e] plus the leaf frequency passed on down-
wards in recursion (rows 16–17).
Let us give a short example using Figure 1(c). As-
sume that we arrive at the range [5,7] which corre-
sponds to subpath P = BA. We branch with B which
gives us the new range [8,8]. However, there is also a
leaf branching with B in the range [5,7] (and recall
that we use lower-case to distinguish leaves), thus,
when we recursively call for the new range [8,8], we
also pass on the information about the branching leaf.
Final frequency of P
0
= BBA is then 2.
The time complexity of each recursion step is
dominated by the range search on the Wavelet tree
(rows 1–2), which requires (with a naive approach)
time proportional to the length of the interval, i.e.
O((e − s + 1)log σ). Since the recursion iterates only
over root-originating paths, the intervals [s,e] are all
non-overlapping. Thus, the total time complexity
is O(t log σ). Moreover, we can generalize the fre-
quency counting for a set of trees or multiple tree sets
while retaining the same time complexity.
Computing the Kernel. We use a Dirac kernel to
compare individual paths. The kernel is computed di-
rectly as a dot product between path feature vectors.
4 EXPERIMENTS
In this section, we evaluate the performance of path
kernels against state-of-the-art graph kernels in a pre-
diction task from (Saigo et al., 2010), where the EC
number of a reaction is predicted. An EC code anno-
tates the function of an enzymatic reaction in a four-
level hierarchical numeric code “a.b.c.d”, where the
first level indicates the general class of the enzyme
(ligases, lyases, isomerases, etc.) and following lev-
els specify the reaction mechanisms. The fourth level
is arbitrary index and ignored. Thus there are 270 full
EC codes.
Reaction Dataset. We perform the experiments
on a reaction set from KEGG LIGAND database
representing common metabolic reactions (release
1.7.2010). A chemical reaction is a graph transforma-
tion, where substrates are transformed into products
often catalyzed by an enzyme, which rearranges the
bonds of the chemical compounds. We constructed
graph representations of the reactions by modeling
the substrates and products as two separate disjoint
graphs with a bi-partite optimal atom mapping be-
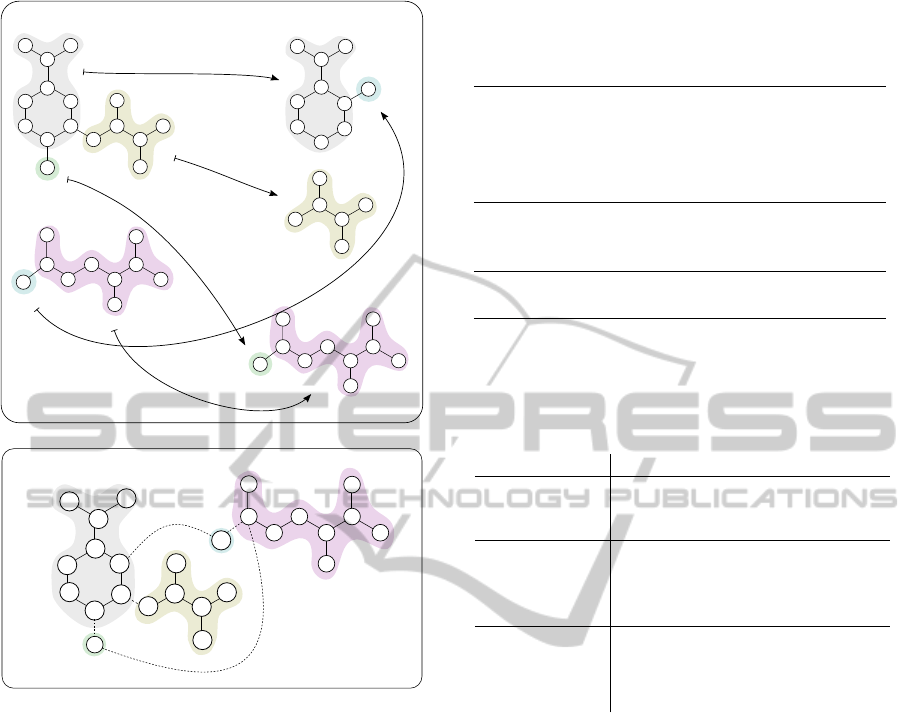
tween the two graphs’ nodes (see top of Figure 3)
(Heinonen et al., 2011). A reaction graph is con-
structed by rerouting edges of the product side to
the substrate side through the mapping, and removing
product nodes (see bottom of Figure 3) (Felix et al.,
2005).
The dataset consists of 15566 EC-labeled reac-
tion graphs computed using optimal atom mappings.
All reactions are theoretically capable of functioning
in forward and backward directions, thus we include
both versions by constructing a direction-invariant
tensor kernel as in (Astikainen et al., 2011) resulting
in 7783 reactions. Median size of reaction graphs is
38 and maximum 393.
Kernels for Reactions. We compare our path ker-
nels to the simple walk kernel (up to length k = 15),
shortest path kernel, and the RGK kernel by (Saigo
et al., 2010), specifically introduced for reactions. In
RGK an alternative reaction graph model is defined,
which uses inner and outer marginal walk kernels to
compare similarity of reactions.
The path index was implemented by reusing our
own existing C/C++ implementations of Huffman-
shaped Wavelet trees (Grossi et al., 2004) and bitvec-
tors supporting rank and select queries (Jacobson,
1989). Optimal construction time and space are not
yet achieved since our current implementation of the
sorting phase requires O(t log
2
t) time and Θ(t logt)
bits of memory. We are currently planning to improve
the sorting phase by plugging in a more sophisticated
path sorting method (Ferragina et al., 2009). Table 1
gives a summary of the performance of the path index.
We ran the experiments with MMCRF hierarchi-
cal multilabel classification algorithm (Rousu et al.,
2007). All kernels use λ = 0.90 and non-RGK ker-
nels are tensor kernels. A 5-fold cross-validation pro-
cedure was used and an optimal C-parameter was cho-
sen from {1,10, 100,1000,10000}. We employ nor-
malized quadratic kernels as they achieved consis-
tently best results.
Results. The results are shown in table 2. A reac-
tion mechanism is deemed correctly classified if the
correct root-to-leaf branch of the EC hierachy is pre-
dicted by MMCRF. The walk kernel is clearly infe-
EFFICIENT PATH KERNELS FOR REACTION FUNCTION PREDICTION
205
N
C
C
C
C
C
O
O
O
N
C
C
C
O
O
O
C
C
C
C
C
C
C
O
O
N
C
C
C
O
O
O
O
C
C
C
C
C
O O
O
N
C C
C C
C
C
O
O
O
C
C
C
C
C
C
C
O
C
C
C
C
O
O
O
O
O
C
C
C
C
C
O
O
O
N
N
new
new
removed
removed
removed
Pyruvate
Anthranilate
Glutamate
Chorismate
Glutamine
Figure 3: Reaction R00986: Chorismate pyruvate-
lyase in forward direction and its optimal atom mapping
(top). Atom mapping is highlighted with corresponding col-
ors and the corresponding reaction graph is below. The “+1”
edges are marked as new and “-1” edges are removed.
rior to the other kernels in this test. RGK and shortest
paths kernels fared better achieving a test set error of
35.0% and 36.4%, respectively. Variants of the path
kernels achieved a minimum of 24.3% test error.
We experimented with upper bounds 15 and 50 for
path length. The results are similar or slightly better
for the upper bound 15, indicating that most informa-
tion resides in shorter paths. However the overfitting
effect of the significantly larger feature space associ-
ated with the bound 50 is relatively small.
With compressed path index it is trivial to pick a
subset of paths to focus on. We experimented with
core paths, paths that go through modified edges (la-
beled “+1” or “-1”). These paths are most likely most
relevant regarding the reaction transformation. The
results show that using core paths decreases the error
to 28.9% from 34.2% of the all path version. How-
ever, when using indicator feature values (all features
are binary) the all-path kernel achieves the lowest er-
Table 1: Characteristics of the test data and performance
results. Note that both construction time and space can still
be optimized with a small engineering effort. Experiments
run on Xeon X7350 CPU and 128 GB of memory.
# of reaction graphs 17,430
# of trees 746,438
# of tree nodes 279 mil.
# of tree leaves 91 mil.
max. tree depth 50
Index construction time 1.1 hours
Index construction space 4.4 GB
Final index size 1.1 GB
# of unique paths 21 mil.
Index frequency computation 176 s
Kernel computation (path length 50) 718 s
MMCRF run (average, 5-fold cv) 10 hours
Table 2: Prediction of full EC class. Core path kernel only
includes paths with “+1” or “-1” edges, while indicator ker-
nels contain only binary values.
Kernel k Tr. error (%) Ts. error (%)
Walk 15 52.9 61.1
RGK inf 27.8 35.0
Shortest paths 21.5 36.4
Core paths 50 14.9 28.9
Core paths, ind 50 14.5 27.8
All paths 50 19.6 34.2
All paths, ind 50 9.1 25.6
Core paths 15 15.0 28.3
Core paths, ind 15 14.7 27.3
All paths 15 20.0 33.7
All paths, ind 15 9.2 24.3
ror rates independent of the upper bound on the path
length. It seems that core paths provides a clearer sig-
nal to the learning machine, but using indicators for
all paths achieves an even stronger effect.
An experiment with the max kernel had a small
less than 1% positive effect on accuracies (data not
shown). The max kernel is not positive semi-definite,
although the smallest negative eigenvalue is relatively
small. The MMCRF seemed to converge in spite of
the indefinite setting. The values on Table 2 are from
the max kernel.
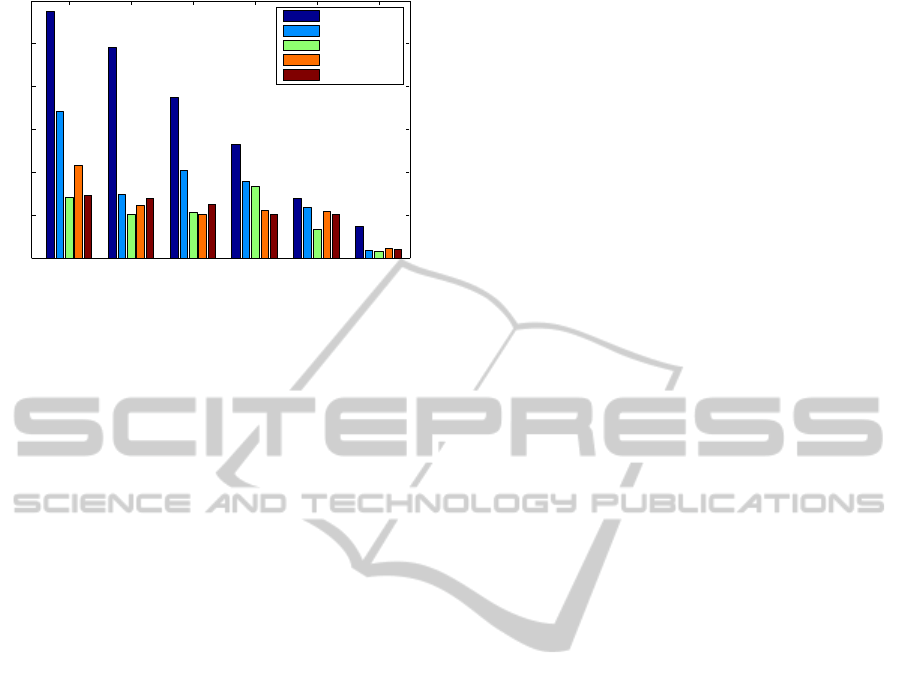
Figure 4 shows the performance of five main ker-
nels on the EC main class only. The main classes are
most general and are thus most difficult to predict.
Here, RGK kernel is on par with path kernels. The
first EC class of oxidoreductases is most difficult to
predict. The high performance on the sixth class of
ligases can be explained by the homogeneity of the
ligases.
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
206
EC1 EC2 EC3 EC4 EC5 EC6
0
2.5
5
7.5
10
12.5
15
EC Class
Test error (%)
Walks
Shortest paths
RGK
Core paths
All paths
Figure 4: Prediction results for the six main EC classes. The
first EC class (oxidoreductases) is most difficult, while the
last (ligases) is well predicted with any method.
5 DISCUSSION AND
CONCLUSIONS
The path index can also be used for on-line path fre-
quency queries: given any path P of length m ≤ h,
where h is the maximum path length enumerated, we
can output the frequency of the path P in all graphs
in just O(m log σ + output) time, where output is the
size of the output, i.e. the number of distinct graphs
having frequency that is greater than zero. Details are
out of the scope of this paper, but this type of on-line
feature query might be interesting when computing
kernels in iterative manner. Another interesting di-
rection would be to implement feature selection or `
1
regularized learning methods for graph data making
use of the efficient access to features.
We presented a method for efficiently computing
all-paths kernels for graph data. Our approach relies
on computing and storing a single compressed path
index of all graphs, which can subsequently be effi-
ciently queried for the purposes for graph kernel or
feature vector computation. We demonstrate the com-
putational feasibility of the approach by computing a
path index for graph representations of KEGG reac-
tions. Our experiments show that path kernels give
significant improvements over walk kernels in the re-
action mechanism prediction task.
ACKNOWLEDGEMENTS
We are grateful to Petteri Kaski, Mikko Koivisto
and Craig Saunders for insightful discussions. This
work was funded by the Academy of Finland grants
1140727 and 118653, the Graduate School Hecse and
by the PASCAL2 (IST grant-2007-216886).
REFERENCES
Astikainen, K., Holm, L., Pitk
¨
anen, E., Szedmak, S., and
Rousu, J. (2011). Structured output prediction of
novel enzyme function with reaction kernels. In
Biomedical Engineering Systems and Technologies,
pages 367–378. Springer.
Borgwardt, K., Ong, C., Sch
¨
onauer, S., Vishwanathan, S.,
Smola, A., and Kriegel, H.-P. (2005). Protein function
prediction via graph kernels. Bioinformatics, 21:i47.
Demco, A. (2009). Graph Kernel Extension and Exper-
iments with Application to Molecule Classification,
Lead Hopping and Multiple Targets. PhD thesis, Uni-
versity of Southampton.
Felix, H., Rossello, F., and Valiente, G. (2005). Optimal
artificial chemistries and metabolic pathways. In Proc.
6th Mexican Int. Conf. Computer Science, pages 298–
305. IEEE Computer Science Press.
Ferragina, P., Luccio, F., Manzini, G., and Muthukrishnan,
S. (2009). Compressing and indexing labeled trees,
with applications. J. ACM, 57:4:1–4:33.
Ferragina, P., Manzini, G., M
¨
akinen, V., and Navarro, G.
(2007). Compressed representations of sequences and
full-text indexes. ACM Transactions on Algorithms
(TALG), 3(2):article 20.
G
¨
artner, T. (2003). A survey of kernels for structured data.
ACM SIGKDD Explorations Newsletter, 5:49–58.
Grossi, R., Gupta, A., and Vitter, J. S. (2004). When index-
ing equals compression: experiments with compress-
ing suffix arrays and applications. In Proc. 15th an-
nual ACM-SIAM Symposium on Discrete Algorithms,
pages 636–645, Philadelphia, PA, USA. SIAM.
Heinonen, M., Lappalainen, S., Mielik
¨
ainen, T., and Rousu,
J. (2011). Computing atom mappings for biochemical
reactions without subgraph isomorphism. J. Comp.
Biology, 18:43–58.
Jacobson, G. (1989). Succinct Static Data Structures. PhD
thesis, Carnegie–Mellon. CMU-CS-89-112.
Kashima, H., Tsuda, K., and Inokuchi, A. (2003). Marginal-
ized kernels between labeled graphs. In Proc. 20th Int.
Conf. on Machine Learning (ICML), pages 321–328.
Mahe, P., Ueda, N., Akutsu, T., Perret, J.-L., and Vert, J.-P.
(2005). Graph kernels for molecular structure-activity
relationship analysis with support vector machines. J.
Chem. Inf. Model., 45:939–951.
Ralaivola, L., Swamidass, S., Saigo, H., and Baldi, P.
(2005). Graph kernels for chemical informatics. Neu-
ral Networks, 18:1093–1110.
Rousu, J., Saunders, C., Szedmak, S., and Shawe-Taylor,
J. (2007). Efficient algorithms for max-margin struc-
tured classification. Predicting Structured Data, pages
105–129.
Saigo, H., Hattori, M., Kashima, H., and Tsuda, K.
(2010). Reaction graph kernels predict ec numbers
of unknown enzymatic reactions in plant secondary
metabolism. BMC Bioinformatics, 11:S31.
Shawe-Taylor, J. and Christianini, N. (2004). Kernel Meth-
ods for Pattern Analysis. Cambridge University Press.
EFFICIENT PATH KERNELS FOR REACTION FUNCTION PREDICTION
207
