
cswHMM: A NOVEL CONTEXT SWITCHING HIDDEN
MARKOV MODEL FOR BIOLOGICAL SEQUENCE ANALYSIS
Vojtěch Bystrý and Matej Lexa
Faculty of Informatics, Masaryk University, Brno, Czech Republic
Keywords: Bioinformatics, Data-mining, Hidden markov models.
Abstract: In this work we created a sequence model that goes beyond simple linear patterns to model a specific type
of higher-order relationship possible in biological sequences. Particularly, we seek models that can account
for partially overlaid and interleaved patterns in biological sequences. Our proposed context-switching
model (cswHMM) is designed as a variable-order hidden Markov model (HMM) with a specific structure
that allows switching control between two or more sub-models. An important feature of our model is the
ability of its sub-models to store their last active state, so when each sub-model resumes control it can
continue uninterrupted. This is a fundamental variation on the closely related jumping HMMs. A
combination of as few as two simple linear HMMs can describe sequences with complicated mixed
dependencies. Tests of this approach suggest that a combination of HMMs for protein sequence analysis,
such as pattern mining based HMMs or profile HMMs, with the context-switching approach can improve
the descriptive ability and performance of the models.
1 INTRODUCTION
Biological sequences, especially protein sequences,
code for 3-D objects, where even a sequentially
distant position can fold into mutual proximity and
vice versa, consecutive amino acids can have
contrasting functions. As such, protein sequences
contain interleaved gapped patterns of varying size,
as well as long-distance interactions and complex
dependencies that often need to be accounted for.
That makes creating a good statistical model of a
biological sequence extremely difficult.
Many approaches to representations of sequences
by specialized data structures and models have been
tried and are still valid, such as suffix trees
(Bejerano, 2001), regular expressions (Nicolas,
2004) or motif descriptions (Bailey, 2009). Perhaps
the most successful and widely known techniques
are based on hidden Markov models (Karplus,
1998), a class of stochastic models working with the
probability of individual symbols in sequences. The
most commonly used models are the profile hidden
Markov models (pHMM) (Eddy, 1998) where each
hidden state represents a specific position in the
biological sequence. Models where states
correspond to a group of sequence positions also
exist. Such models have been used for
transmembrane protein topology prediction
(Viklund, 2004) or gene finding. (Pachter, 2001)
Lately, more complex variations appeared such as
jumping profile hidden Markov models (jpHMM)
(Schultz, 2006) or the gapped pattern-mining-based
hidden Markov model VOGUE (Zaki, 2010).
VOGUE adds the ability of modelling gapped
patterns and its biggest contribution is the idea of
combing data mining with data modelling. We have
adapted this data mining technique as a part of the
parameter learning process of our model. jpHMM
improve upon simple HMMs by allowing jumping
between different sub-models. That allows jpHMM
to better model alternating contexts in sequences.
Strict adherence to the markovian property causes
the history of visited states to be completely
forgotten after every jump to a different sub-model.
To address this shortcoming, we propose a new
type of hidden Markov model that is a combination
of jumping profile HMMs and variable order
HMMs. Same as jpHMM, our model consists of
distinct sub-models, each representing a different
context in the sequence. Similarly to jpHMM, our
model can arbitrarily switch between these sub-
models. Our model adds a context memory by
remembering the last active state of each sub-model,
208
Bystrý V. and Lexa M..
cswHMM: A NOVEL CONTEXT SWITCHING HIDDEN MARKOV MODEL FOR BIOLOGICAL SEQUENCE ANALYSIS.
DOI: 10.5220/0003780902080213
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2012), pages 208-213
ISBN: 978-989-8425-90-4
Copyright
c
2012 SCITEPRESS (Science and Technology Publications, Lda.)

so that these can continue the computation
uninterrupted after resuming control. It means that
each sub-model can represent different arbitrarily-
gapped context in the sequence and cswHMM can
combine them together. That gives cswHMM a
unique ability to analyze sequences that contain
mixed signals with interleaving patterns.
We envision applications of cswHMM in several
areas of biological sequence analysis. For example,
proteins from a single family or superfamily often
share a common amino acid core, but differ in the
surface amino acids, often located in loops of
varying lengths. While the composition of the core is
driven by the necessity for predictable protein
folding, the external parts may be determined by
possible binding partners, the surrounding
environment or a required enzymatic activity. The
two kinds of sequences alternate in most currently
known protein folds (Fernandez-Fuentes, 2010),
therefore modelling such sequences with cswHMM
seems natural. Another candidate for the application
of cswHMM may be found in gene prediction. The
position of genes in nucleotide sequences is often
predicted using HMMs. In the case of eukaryotic
genes, part of the HMM predicts exons interleaved
with introns. Because the exons contain 3-nucleotide
codons which must remain in-frame even between
two exons separated by an intron, successful
prediction of precise exon/intron boundaries requires
a memory mechanism identical to the one proposed
in cswHMMs (Majoros, 2009). Membrane proteins
often form interfaces between two environments
separated by the membrane, the parts of the protein
that form one or the other part of such interface are
commonly interleaved in the primary sequence
(Krogh, 2001), again giving a possibility to model
such sequences with cswHMM better than with
alternative models. The other broader goal is to
identify new gapped motifs in protein sequences and
the possibility of their combination. This will allow
us to contribute to a better understanding of protein
sequence composition (Ganapathiraju, 2005).
The rest of the paper is organized as follows. In
Section 2 we describe in detail the general design of
cswHMM and its formal definition. In the end of the
section we show a possible way of unsupervised
learning of the cswHMM based on mined gapped
patterns. In Section 3 we present two applications of
our model to protein sequence analysis. Finally,
Section 4 concludes our work and presents possible
future research.
2 METHODOLOGY
HMM is the mathematical basis for our model since
cswHMM is a case of a HMM with special
topology. To summarize and unify the notification
we will briefly describe the formal definition of
HMM.
2.1 HMM Definition
HMMs are defined by a set of discrete hidden states
=
{
…
}
a transition function (
,
)=
providing
the probability of
transition from state
to state
; by its output
density probability function (
)=
(
|
) where
(
|
) is a random variable over the set of possible
observations determining probability of these
observations. For applications in sequence analysis
the set of possible observations is usually equal to
sequence alphabet and it is a discrete variable. The
last thing to define in HMM is a vector of prior
probabilities. In the following text we omit
definitions of these, since they are irrelevant to our
model and can always be replaced by an additional
initial state and a corresponding addition to the
transition function.
2.2 cswHMM Definition
As explained in Section 1, the basic idea of
cswHMM is to combine different sub-models so
they can transit from one to another while keeping
the last active state in all other sub-models in a
specialized memory. A classical hidden Markov
model is memoryless, but we can encode the
information about the last active states in the
topology of the HMM.
Figure 1: a) Transition diagram of two HMMs we want to
join. b) Transition diagram of joined cswHMM.
To join two HMM sub-models with a set of states X
and Y, we create a model with two sets of states,
each composed of the Cartesian product of X and Y.
Each state of cswHMM can be identified by an
cswHMM: A NOVEL CONTEXT SWITCHING HIDDEN MARKOV MODEL FOR BIOLOGICAL SEQUENCE
ANALYSIS
209

ordered triplet ,
,
where ∈{1,2} is the
number of the currently active sub-model.
∈
and
∈ are the last active states in the
respective sub-models. Figure 1 illustrates how two
simple HMMs are joined to cswHMM. In the next
paragraph we present the formal definition of
cswHMM for joining of n sub-models. We define
the emission and transition probability functions of
such model.
Suppose we have a set of HMMs Λ=
{
H
…H
}
, where each
has states
=
…
, transition probability function
(
,
)=
), and output probability
density function
(,
)=
(
|
), where
,∈
{
1…
}
and o are possible observation. Let
there be an index set =
{
1…
}
. Then cswHMM
created as a combination of individual HMMs from
Λ would have states S according to Equation 1.
Equation 2 defines the emission probability function
O and the transition probability function T of such
cswHMM is defined by Equations 3-5.
=×
×
×…×
=
=
…
{
,
,…,
}
(1)
,
,…,
=
(2)
,
,…,
,…,
,
,
,…,
,…,
=
,
(3)
,
,…,
,…,
,
,
,…,
,…,
:≠
=
,
∗
,
(4)
otherwise
(
,
)
=0 (5)
Where
(
,
)
in the equation (4) is the
probability of switching from state j of sub-model H
i
to sub-model H
l
. Essentially, parameter
(
,
)
reflects the frequency of switching between
contexts. Since we may not know the mapping of
contexts on the data until the end of the learning
procedure, estimation of this parameter is
problematic and has to be done iteratively during the
learning process or we must use some known
properties of the data.
With this topology the cswHMM can switch
between its underlying sub-models without losing
knowledge of the continuity in the sub-models. That
makes cswHMM a powerful tool for modelling data
with interleaving patterns.
2.3 State Space Reduction
The drawback of such a general model is its
complexity, since cswHMM composed of n HMM,
each with m states ∗
has states. Since the
algorithms for inference of HMM have time
complexity Ο=
(
∗
)
where N is the number of
states and L is the length of analyzed sequence, the
time complexity of inference for such a cswHMM
would be Ο=
(
∗
∗
)
. It is therefore
necessary to build cswHMMs from a small number
of relatively small sub-models in order to keep the
models computationally feasible.
In real-world problems it is not usually necessary
to combine all states of every sub-model, since in a
linear sequence the number of directly neighbouring
and possibly interleaved contexts is limited. The
following methods can be used to lower the state
complexity of cswHMM. If there are set of states of
individual sub-model which do not switch to other
sub-models, we can model such sub-models as a
hierarchical HMM, where the “non-switching” set of
states will be encapsulated in one higher-level state.
The switching will take part only between these
higher-level states.
The second method to lower state complexity is
to combine only the sub-models that might
interleave in the sequence. If we had two sub-models
that we know will not mix in the sequence, we can
create two separate cswHMMs, each with only one
such model and then connect these cswHMM in
parallel via a general state. This method allows us to
create one complex model of a protein family that is
made of many relatively small cswHMMs. Thus, the
overall state complexity of the model is
computationally feasible.
To conclude this section, let us emphasize that
cswHMM may itself be a deep and complex model,
but it is always a result of combining simpler
HMMs. As such, its overall quality always depends
on the sub-models we use.
2.4 Learning cswHMM
As mentioned before, the combination of models
and the lack of precise knowledge about switching
between them makes it difficult to learn model
parameters directly from data. For HMMs this is
commonly done by some type of EM algorithm.
Inability to directly separate training data into sub-
models requires a different approach. A possible
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
210
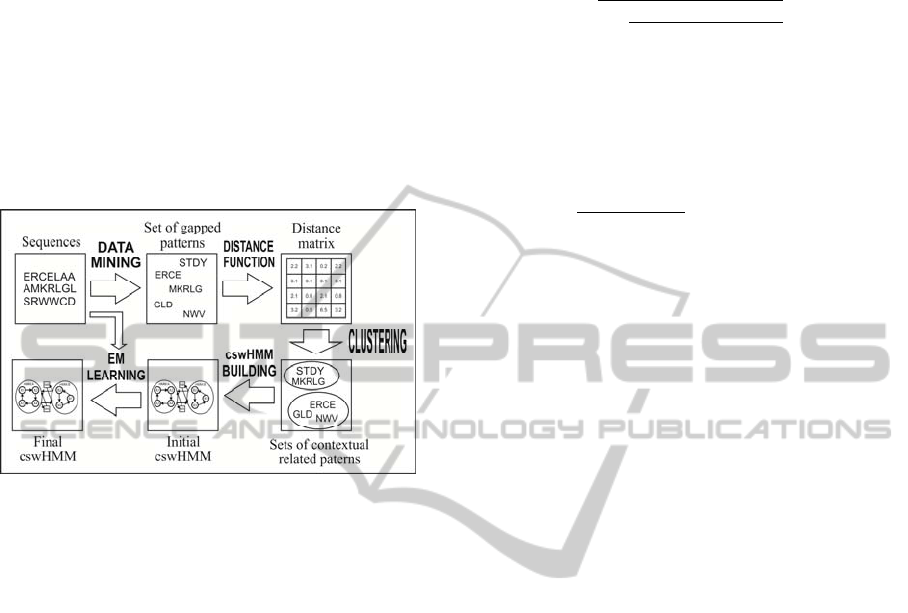
solution is to use data mining to estimate the
separation of data into sub-models and a subsequent
data modelling step based on the mined patterns. A
similar technique of combining data mining and data
modelling has been used in VOGUE (Zaki, 2010),
we therefore used its backbone as a basis for our
algorithm. Figure 2 shows the general scheme of the
necessary algorithms. The main difference in our
approach is the separation of patterns using
clustering to obtain more models for different
contexts. Next, we will describe individual parts of
our algorithm in detail.
Figure 2: General diagram of cswHMM learning process
via mined patterns.
We used pattern mining algorithm VGS used as a
base for the VOGUE model, which itself is a
modification of cSPADE (Zaki, 2001), a method for
constrained sequence mining. VGS finds all patterns
of length at most k, with the maximum gap g
between each two elements that have minimal
frequency f in the sequence. Variables ,, are
parameters of the algorithm set by the user. VGS
starts with patterns of lengths k = 1 and then
incrementally couples those with enough positions
in mutual proximity (defined by g) to reach
frequency . As an output of pattern mining we have
a set of relevant patterns, each consisting of its ID
and a list of positions of patterns occurring in the
sequence.
In the second step of our algorithm we compute a
similarity matrix in order to cluster mined patterns in
the different sets representing individual contexts in
the sequences. The logic behind “similarity” of two
patterns is that if they frequently overlap and extend
each other it is likely that they belong to the same
context and therefore to the same pattern set. On the
other hand patterns which frequently interleave and
mix without coinciding with each other are more
likely to belong to different contexts. We therefore
define a similarity function
,
→[0,1] in
Equation 7 where
and
are patterns we want to
compare.
,
=
1
1+
max
(
0,
+⋯+
)
(6)
Here n is the number of instances of
and
overlapping in the sequence.
,…,
are respective
distance coefficients of these collisions and is a
coefficient determining the steepness of the function.
Distance coefficients
are defined by Equation 7.
=
+
2
−∗
(7)
where
is the number of elements of pattern
that lie in some gap of pattern
. Similarly,
denotes the same for elements of
in some gap of
. is the number of elements of patterns that
coincide and
and
are weights set by the user.
For the clustering itself we used direct clustering
as implemented in the clustering tool CLUTO
(Karypis, 2002), since direct clustering is the best
clustering method for small number of clusters. Each
cluster represents one set of patterns that in turn
represent one context in the sequence.
Individual HMMs which are the bases for our
cswHMM are created from each pattern set similarly
to the VOGUE algorithm, except we don’t add the
gap states in the individual HMMs. Instead we add
an additional HMM consisting of a single gap state,
representing the parts of sequence not covered by
any other HMMs. cswHMM is consequently created
by combining these HMMs as described in Section
2.
Once we obtain an estimation of the appropriate
cswHMM, we can separate data into sub-models and
fine-tune our model using common EM methods,
such as the popular Baum-Welsch algorithm.
3 VALIDATION AND POSSIBLE
APPLICATIONS
3.1 PROSITE Classification
With this type of unsupervised learning we tested
the performance of cswHMM as applied to the
problem of protein classification. To compare our
model with other existing HMMs, we chose the
same dataset and type of experiment as the authors
of the VOGUE (Zaki, 2010) model.
From the dataset of protein families PROSITE
(Nicolas, 2004), ten families were chosen as a
cswHMM: A NOVEL CONTEXT SWITCHING HIDDEN MARKOV MODEL FOR BIOLOGICAL SEQUENCE
ANALYSIS
211

testing dataset. Each family was divided into a
training and testing set. Precisely, 75% of family
members were used as training data for individual
models, while remaining 25% of each family was
compiled into a classification testing set.
Table 1: Accuracy of protein classification for individual
protein families from PROSITE and overall accuracy.
Class
cswHM
M
VOGUE HMMER HMM
PDOC00662 81,82 81,82 72,73 27,27
PDOC00670 85,71 80,36 73,21 71,4
PDOC00561 90,48 95,24 42,86 61,9
PDOC00064 85,71 85,71 85,71 85,71
PDOC00154 71,88 71,88 71,88 59,38
PDOC00224 91,67 87,5 100 79,17
PDOC00271 91,89 89,19 100 64,86
PDOC00343 92,85 89,29 96,43 71,43
PDOC00397 80 100 40 60
PDOC00443 85,71 100 85,71 85,71
Average 86,38 85,11 80,43 67,66
We trained the cswHMM with different values of
parameters of maximal pattern length, maximal gap
length, minimal pattern frequency, similarity
function coefficients and the switching probability.
The models that gave the best performance were
chosen. The optimal value of maximal pattern length
was found to be 6 to 7. The maximal gap length was
found not to significantly influence the results if
higher than 5. The average probability of switching
between sub-models was 0,86. This means that most
of the modelled patterns were gapped.
Table 1 shows the comparison of results of
different models on individual datasets and the
overall probability of prediction. cswHMM is
comparable with other methods and in overall
probability it even slightly surpass them, but this
type of application should not be the primary
function of cswHMM. We use it more as a
validation of our proposed model whose main
purpose is to improve analyses of sequences with
mixed contexts and to identify those contexts. To do
so, we currently develop a tool to analyze individual
sub-models and their performance during sequence
analysis.
3.2 Loop Modelling
We analyzed eight arbitrarily selected protein
families defined in the Pfam database (Finn, 2010),
using the alignment of seed sequences of each
family to determine conserved (protein core) and
variable (loops) regions. In each alignment we
identified possible loop positions as positions where
more than 30% of aligned sequences had a gap.
Amino acids at these positions were used to create a
database of short sequences that represent the
possible loops.
Table 2: Logarithmic probabilities for combined HMM,
classic profile HMM and their comparison.
Pfam code cswHMM pHMM difference %
PF00078 -3,738 -3,670 0,07 1,84
PF00024 -3,856 -3,756 0,10 2,66
PF00117 -3,722 -3,709 0,01 0,34
PF00171 -3,834 -3,798 0,04 0,95
PF00227 -3,699 -3,685 0,01 0,39
PF00246 -3,786 -3,695 0,09 2,46
PF03129 -3,856 -3,752 0,10 2,77
PF01436 -3,864 -3,812 0,05 1,36
Average -3,794 -3,735 0,060 1,60
We trained a four-state HMM on this database to
create a simple loop model for each family.
Subsequently, we used the core profile HMM of
each family and combined it with the corresponding
loop model to create a simple cswHMM. We
computed logarithmic probabilities of generating
individual family without insertion states sequences
with the new model and compared them with the
logarithmic probabilities for classical pHMM. The
results are shown in Table 2 and show slight
improvement of generating probability with
cswHMM.
The loop modelling experiment has shown that
the cswHMM approach may find use in protein
sequence analysis where blocks of amino acids
interfacing with different environments (protein
core, membrane or cytoplasm) are interspersed with
contrasting blocks. Numerical treatment
corresponding to the proposed cswHMM provided
an average 1,6% improvement in sequence
description, consistently better for all studied
families (Table 2). Models used within the
cswHMM framework could possibly represent
different building blocks, where at least one of the
block types follows a predetermined order of amino
acids and where mixing of the blocks is variable or
optional.
4 CONCLUSIONS AND FUTURE
WORK
In this paper we have presented a new model that is
a type of variable-order hidden Markov model with
ability to analyze mixed contexts in sequences. We
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
212

also introduced one possible method for
unsupervised learning of cswHMM based on mined
gapped patterns and showed two possible
applications of such model to protein sequence
analysis.
In our future work we plan to apply our
cswHMM to identify other mixed contexts in
biological sequences. The possible application area
is twofold. We can use our model to combine
existing models of sequences that are known to have
non-uniformly mixed contexts and in that way
increase the description ability of those models.
Some examples of such sequences are in Section 1.
The other way of application of cswHMMs is to use
unsupervised learning over different sets of
sequences and try to find new, currently unknown
common gapped patterns and their possible
combinations. In that way we could reveal new rules
governing biological sequence composition.
ACKNOWLEDGEMENTS
This work has been supported by the Grant Agency
of the Czech Republic grant GD204/08/H054.
REFERENCES
Bailey, T. L., et al., 2009. MEME SUITE: tools for motif
discovery and searching, Nucl. Acids Res. 37(suppl 2)
Bejerano, G., Yona, G., 2001. Variations on probabilistic
suffix trees: statistical modeling and prediction of
protein families Bioinformatics 17(1): 23-43
Eddy, S. R., 1998. Profile hidden Markov models.
Bioinformatics 14(9): 755-763
Fernandez-Fuentes, N., Dybas, J. M., Fiser, A., 2010.
Structural Characteristics of Novel Protein Folds.
PLoS Comput Biol 6(4)
Finn R. D., et al., 2010. The Pfam protein families
database. Nucleic Acids Research, Database Issue 38:
D211-222
Ganapathiraju, M., et al., 2005. Computational Biology
and Language.
Karplus, K., Barrett, C., Hughey, R., 1998. Hidden
Markov models for detecting remote protein homolo-
gies. Bioinformatics 14(10): 846-856
Karypis, G., 2002. CLUTO a Clustering Toolkit, Techni-
cal Report 02-017, Dept. of Computer Science, Univ.
of Minnesota, http://www.cs.umn.edu/cluto
Krogh, A., Larsson, B., von Heijne, G., Sonnhammer, E.
L., 2001. Predicting transmembrane protein topology
with a hidden Markov model: Application to complete
genomes. JMol Biol 305:567–580.(2001)
Majoros, W. H., Korf, I., Ohler, U., 2009. Gene Prediction
Methods Bioinformatics, 99-119,
Nicolas, H. et al., 2004. Recent improvements to the
PROSITE database Nucl. Acids Res. 32(suppl 1):
D134-D137
Pachter L., Alexandersson M., Cawley S., 2001.
Applications of generalized pair hidden Markov
models to alignment and gene finding problems
(RECOMB '01). ACM, New York, NY, USA, 241-248.
Schultz, A. K., et al., 2006. A jumping profile Hidden
Markov Model and applications to recombination sites
in HIV and HCV genomes, BMC Bioinformatics 2006,
7:265
Viklund H., Elofsson A., 2004. Best α-helical
transmembrane protein topology predictions are
achieved using hidden Markov models and
evolutionary information Protein Sci. 13(7): 1908–
1917.
Zaki, M. J., Carothers, Ch. D., Szymanski, B. K., 2010.
VOGUE: A variable order hidden Markov model with
duration based on frequent sequence mining. ACM
Trans. Knowl. Discov. Data 4, 1, Article 5 (January)
Zaki, M. J., 2001. SPADE: An efficient algorithm for
mining frequent sequences. Mach. Learn. J. 42, 1/2,
31–60.
cswHMM: A NOVEL CONTEXT SWITCHING HIDDEN MARKOV MODEL FOR BIOLOGICAL SEQUENCE
ANALYSIS
213
