
Predicting Molecular Functions in Plants using Wavelet-based Motifs
G. Arango-Argoty
1
, A. F. Giraldo-Forero
1
, J. A. Jaramillo-Garz´on
1,2
, L. Duque-Mun˜oz
1,2
and G. Castellanos-Dominguez
1
1
Signal Processing and Recognition Group, Universidad Nacional de Colombia,
Campus la Nubia, Km 7 v´ıa al Magdalena, Manizales, Colombia
2
Grupo de M´aquinas Inteligentes y Reconocimiento de Patrones - MIRP, Instituto Tecnol´ogico Metropolitano,
Cll 54A No 30-01, Medell´ın, Colombia
Keywords:
Amino Acid Properties, Dissimilarity based Classification, Molecular Function, Motifs, Wavelet Transform.
Abstract:
Predicting molecular functions of proteins is a fundamental challenge in bioinformatics. Commonly used
algorithms are based on sequence alignments and fail when the training sequences have low percentages of
identity with query proteins, as it is the case for non-model organisms such as land plants. On the other
hand, machine learning-based algorithms offer a good alternative for prediction, but most of them ignore that
molecular functions are conditioned by functional domains instead of global features of the whole sequence.
This work presents a novel application of the Wavelet Transform in order to detect discriminant sub-sequences
(motifs) and use them as input for a pattern recognition classifier. The results show that the continuous wavelet
transform is a suitable tool for the identification and characterization of motifs. Also, the proposed classifi-
cation methodology shows good prediction capabilities for datasets with low percentage of identity among
sequences, outperforming BLAST2GO on about 11,5% and PEPSTATS-SVM on 16,4%. Plus, it offers major
interpretability of the obtained results.
1 INTRODUCTION
Functions of gene products are specified by the
molecular activities they perform. These functions
may include transporting other molecules around,
binding to different compounds or holding molecules
together for fastening reactions. Several computa-
tional methods for protein function prediction use se-
quence alignment tools such as BLASTP (Johnson
et al., 2008), which are designed to transfer functions
from already annotated sequences to the novel ones
based on sequence similarity criteria (Cheng et al.,
2005). In this matter, homologous proteins can be
identified under the assumption that amino acids hav-
ing an important role in protein function and struc-
ture cannot mutate without an important effecton pro-
tein activity. However, those amino acids can change
very slowly in a given protein family during evolu-
tion (Liu et al., 2006) and thus, for a set of sequences
that stretch a great evolutionary distance, it is possible
to highly conserved amino acid regions, even if they
greatly differ from a global perspective. On the other
hand, when the sequence similarity is low, aligned
segments are often short and occur by chance, lead-
ing to unreliable and unusable alignments when the
sequences have less than 40% and 20% similarity, re-
spectively (Cheng et al., 2005).
Recently, a vast number of predictors based on
pattern recognition techniques have been designed in
an effort to find alternative methods that do not rely
solely on alignments. Each one of them computes a
different set of attributes to characterize protein se-
quences, including statistical and physical-chemical
properties of amino acids (Shen and Burger, 2010),
energy concentrations from time-frequency represen-
tations (Gupta et al., 2009), distance measures, word
statistics, Hidden Markov Models, information the-
ory and others (Vinga and Almeida, 2003). However,
most of them only describe global attributes of the
whole protein sequence, ignoring the fact that func-
tional domainsmay reside in different portions of pro-
teins within the same family. Such recurring patterns
are called MOTIFS and they can be used to identify
representative regions of the proteins, revealing po-
tential information about their molecular function.
Nevertheless, only a small portion of proteins have
clearly identifiable sorting signals in their sequence
and, since proteins are commonly able to perform sev-
eral molecular functions instead of only one, there
is a strong challenge on how to use those motifs for
140
Arango-Argoty G., F. Giraldo-Forero A., A. Jaramillo-Garzón J., Duque-Muñoz L. and Castellanos-Dominguez G..
Predicting Molecular Functions in Plants using Wavelet-based Motifs.
DOI: 10.5220/0004234201400145
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2013), pages 140-145
ISBN: 978-989-8565-35-8
Copyright
c
2013 SCITEPRESS (Science and Technology Publications, Lda.)

predicting molecular functions with the less possible
amount of false positives and false negatives.
The Wavelet Transform (WT) has been previously
used as a powerful tool for mining information in pro-
teins (Murray et al., 2002). Here, a novel applica-
tion of the WT is developed, extending the represen-
tation scheme to a complete classification methodol-
ogy. First, a protein is decomposed into a set of sub-
sequences by means of the WT. These sub-sequences
are further clustered to build a set of prototype motifs
representing the original protein sequence set. Proto-
type motifs are then used as features in order to build
a representation space, and hence being able to infer
classification rules based on pattern recognition tech-
niques. The properties of the proposed method are:
I) detection of variable length motifs; II) identifica-
tion of patterns distributed in any position along the
sequences and and III) accurate prediction of protein
molecular functions including proteins associated to
multiple functions.
2 MATERIALS AND METHODS
2.1 Experimental Setup
The proposed methodology is depicted in Figure 1. In
step 1, the supervised training set of proteins (molec-
ular function) is preprocessed to extract short sub-
sequences of variable length. These patterns are de-
termined by interactions among adjacent amino acids
represented by wavelet coefficients. In step 2, all the
extracted sub-sequences are clustered to get the pro-
totype motif set. Due to the variable motif length,
the multiple sequence alignment is used to compute
the consensus of all sub-sequences belonging to one
cluster. In step 3, a new protein sequence can be rep-
resented as the minimum distance between the pro-
totypes and its own sequence motifs. Once all pro-
teins are mapped into the set of prototype motifs, a
Support Vector Machine classifier is trained to predict
their molecular function.
All experiments are carried out on land plants
(embryophyta) proteins, belonging to nine differ-
ent molecular functions, as shown in Table 1. A
dataset of 1008 Embryophyta proteins is reported by
UNIPROT
(Jain et al., 2009) (file version:
24-01-11
),
with, at least, one annotation in the ontology
molecular function of Gene Ontology Annotation
Project (Barrell et al., 2009) (file version:22-12-10)
and whose evidence of existence is neither unknown
nor predicted by computational tools. To avoid bias
due to the presence of protein families, the database
does not contain protein sequences with a pair-wise
Figure 1: Main methodology a) The sequences are con-
verted into numerical signals and the CWT is applyed to
obtain two-dimensional representations (position in the x-
axis and amino acid interaction in the y-axis). Detected
motifs are marked with numbers. b) Clustering of detected
motifs and logos representation. c) The distance between a
query protein and the prototype motifs is used to train/test
the classifier.
Table 1: Number of protein sequences per class.
Functions Entire Reduced Functions Entired Reduced
NtBind 109 53 Transp 280 133
TranscFact 160 102
LipBind 38 24
RnaBind 80 52
Kinase 224 103
Nase 33 24
Enzreg 78 46
RecepBind 40 27
similarity superior to 40%. The web server version
of cd-hit (Huang et al., 2010) is used to filter the
dataset by similarity; the remaining number of se-
quences obtained after this process is 564. Classes are
defined according to the GO Slim Classification for
Plants (Swarbreck et al., 2008).
2.2 Extraction of Motifs
Let S = {s
s
s
i
}, i = 1, 2,...,M, be the training set of pro-
tein sequences. Then, a given protein s
s
s
i
of length n
i
can be represented as a numerical signal η
i
(t) that is a
function of its length, by substituting each amino acid
with its equivalent value of a given physical-chemical
propertyI . After all proteins havebeen convertedinto
the numerical signal set η
η
η = {η
i
(t)}, they are pro-
jected by the Continuous Wavelet Transform (CWT)
that is defined as the decomposition of a signal η
i
(t),
as follows:
W
W
W
η
i
(a,b) =
1
p
|a|
!
∞
Z
−∞
η
i
(t)ϕ
t − b
a
dt, (1)
where ϕ((t − b)/a) is the basis wavelet function at a
particular scale a and a translation b, with a,b ∈ R,
a ≥ 0. This work uses the Gauss mother wavelet due
to its smoothing property (Murray et al., 2002).
PredictingMolecularFunctionsinPlantsusingWavelet-basedMotifs
141

The resulting matrix W
W
W
η
i
∈ R
n
S
×n
i
, is called
“scalogram”, and n
S
represents the maximum scale
(or motif length) considered for the decomposition.
It has been empirically fixed to provide an acceptable
trade off between time complexity and maximum mo-
tif length to n
S
= 64. W
W
W
η
i
provides the localization
of frequent sub-sequences within a given sequence s
s
s
i
.
Particularly, for regions with a similar amino acid be-
havior along the sequence, i.e., having high energy
concentrations, it is possible to locate the centroid
point in the scale-position space. Then, this point
grows in the position axis towards both the left and
the right sides until the value of the actual position
becomes less than the minimal value of the region,
and therefore, determines the respective set of n
j
mo-
tifs for the sequence s
s
s
i
, {ξ
ξ
ξ
ij
: j = 1,. .., n
j
}⊂s
s
s
i
,. This
process is applied to each sequence in S .
Regarding the physical-chemical property I , used
for converting sequences into numerical signals, a
total of 51 indexes was selected from the
AAINDEX
database (Kawashima and Kanehisa, 2000). Such in-
dexes involve the six regions of the amino acid prop-
erties, aiming to explore different numerical represen-
tations.
2.3 Dissimilarity Space Representation
In order to obtain representative motifs within the k-
th labeled class, motif subsequences are clustered by
using the well known Iterative Self Organizing Data
Analysis Technique (
ISODATA
). For the implementa-
tion of the algorithm, the alignment-score distance
d(·,·) ∈ R
+
, between any two motifs ξ
ξ
ξ and ν
ν
ν is de-
fined as follows (subscripts are ignored since the orig-
inal sequnece of each motif is irrelevant in this con-
text):
d(ξ
ξ
ξ,ν
ν
ν) =
1−
˜
d(ξ
ξ
ξ,ν
ν
ν)
˜
d(ξ
ξ
ξ,ξ
ξ
ξ)
1−
˜
d(ξ
ξ
ξ,ν
ν
ν)
˜
d(ν
ν
ν,ν
ν
ν)
, (2)
where
˜
d(·,·) is the similarity between two sequences
ξ
ξ
ξ and ν
ν
ν computed as:
˜
d(ξ
ξ
ξ,ν
ν
ν) =
n
ξ
∑
l=1
D
D
D(ξ
ξ
ξ(l),ν
ν
ν(l)) (3)
being n
ξ
the minimal length of both subsequences un-
der consideration, and D
D
D(ξ
ξ
ξ(l),ν
ν
ν(l)) the value of the
scoring matrix for the respective l-th elements of ξ
ξ
ξ
and ν
ν
ν. As scoring matrix D
D
D, the Point Accepted Mu-
tation (
PAM250
) is used for the pairwise local align-
ment, as recommended in (Wheeler, 2002).
The
ISODATA
algorithm produces a set of n
k
C
clus-
ters for each class. Then, as stated in (Schnei-
der, 2002), one prototype motif ζ
ζ
ζ
k
r
, r = 1,... ,n
k
C
, is
generated as the consensus sequence of each cluster.
Given the profile matrix P
P
P
k
r
with elements P
P
P
k
r
(i, j) =
f
k
(i, j)/kC
k
r
k, where f
k
(i, j) represents the cardinal
of amino acid j at position i of the multiple sub-
sequence alignment C
k
r
, then, each component of the
consensus sequence is computed:
ζ
ζ
ζ
k
r
( j) = max
∀i
{P
P
P
k
r
(i, j)}, (4)
Once the set of prototype motifs {ζ
ζ
ζ
k
r
} has been
computed, a query protein z
z
z can be represented by
the minimum alignment-scoredistances between such
prototype motifs and its own motifs ξ
ξ
ξ
i
. The scalar-
valued r-th component of the feature space represen-
tation is computed as:
δ
r
= min
∀ξ
ξ
ξ
i
∈z
z
z
{d(ξ
ξ
ξ
i
,ζ
ζ
ζ
r
)}, r = 1,2, ... ,n
C
(5)
where n
C
=
∑
k
n
k
C
. Conceptually, quantity δ
r
∈ R
+
is
a measure of the extent at which the prototype motif
ζ
ζ
ζ
r
is present in the sequence z
z
z.
2.4 Classification Methodology
The entire database is divided into modeling and
classification sets in which, the 60% of the se-
quences are selected to compute the prototype motifs,
whereas 40% are left for testing purposes. The Fast
Correlation-Based Filter (FCBF), described in (Yu
and Liu, 2003), is used for feature selection. Since
basic SVM are designed only for two-class prob-
lems, classification is implemented following the one-
against-all strategy, which produces a strong class im-
balance. So, the Synthetic Minority Over-sampling
Technique is employed (Chawla et al., 2002). Param-
eters of the SVM are tuned with a Particle Swarm Op-
timization algorithm. Validation of the results is ob-
tained by 10-fold cross-validation over the testing set
(40%). Sensitivity (S
n
), specificity (S
p
), and geomet-
ric mean (G
m
) are used as classification performance
measures:
S
n
=
n
TP
n
TP
+n
FN
S
p
=
n
TN
n
FP
+n
TN
G
m
=
p
S
n
S
p
where n
TP
,n
FP
,n
TN
, and n
FN
denote true positive,
false positive, true negative and false negative, respec-
tively.
2.5 Comparison with other Methods
Blast2GO
: is a research tool designed with the main
purpose of enabling Gene Ontology (GO) based data
mining on sequence data for which the GO an-
notations are not available. Annotation based on
Blast2GO
is carried out by three sequential stages,
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
142
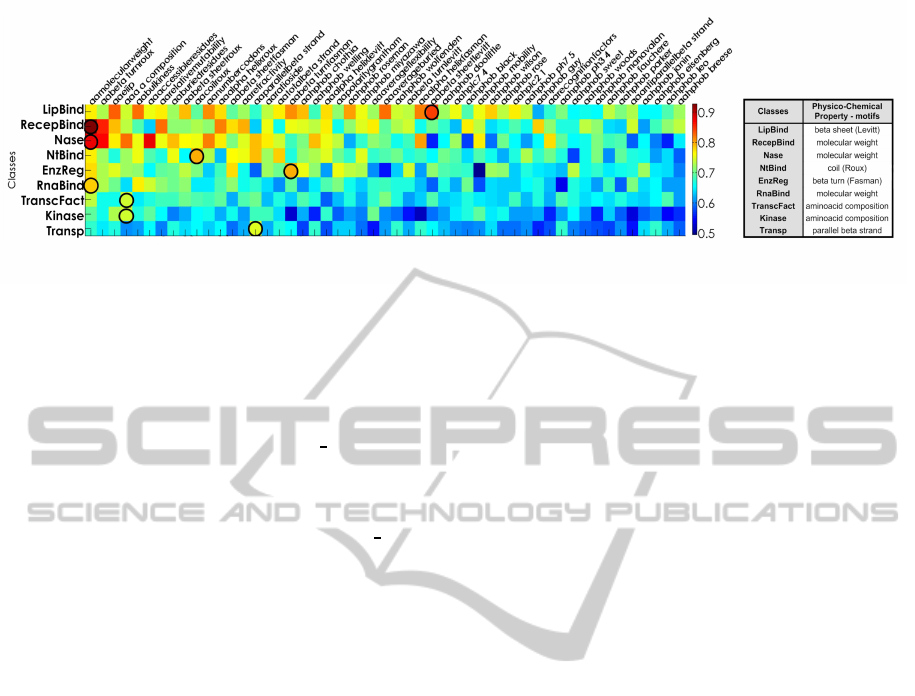
Figure 2: 1) classification performance (geometric mean) for several amino acid properties. 2) selected properties and perfor-
mance of the ensemble prototype motifs.
Blasting, mapping and annotation. For Blasting
the
BLASTP
algoritm is trained and tested over the
same database, holding the same validation method-
ology described in section 2.4. For this purpose,
the
Blast+ version 2.2.26
software is used (pa-
rameters: blosum 62, e-value 10, word size >= 2 -
outfmt 5). In the mapping stage, BLAST results are
loaded to BLAST2GO module (-E-Value-Hit-Filter
10 -Annotation CutOff 10 to improve the false posi-
tive rate) in order to map these results to b2g jun11
database. Finally, in the annotation stage the test-
ing sequences are labeled using the evidence code
weights proposed by
Blast2GO
(Conesa and G¨otz,
2008).
Pepstats-SVM
: is a pattern recognition approach
that uses 37 global features proposed in
Pepstats
(Sarac¸, 2010). The same classification framework
used in section 2.4 is applyed for comparisson pur-
poses. The goal of this comparisson is to show that,
under the same conditions, the prototype motif based
method overpasses the performance of methods based
on global features.
3 RESULTS AND DISCUSSION
Figure 2 depicts the prediction performance using 51
amino acid properties from
AAINDEX
database. Lipid
binding proteins are diverse in sequence, structure,
and function (Lin et al., 2006), so, Lipid binding
proved to be the molecular function that showed the
highest performance within the whole set amino acid
properties.
Receptor binding proteins interact selectively
with one or more specific sites on a receptor
molecule (Lodish et al., 1995). Protein receptors
are transmembranal proteins whose conformation is
given by α, β structures, and some specific domains
(DNA-binding domains, hormone-binding domain,
transmembrane subunits among others). A clear in-
fluence between structure of the receptor proteins and
β-turn and α-helix properties was evinced.
Nucleases are enzymes that participate in nucleic
acid catabolism and play roles in DNA replication,
cutting DNA molecules into small fragments (en-
donuclease activity) and DNA repair by proofreading
(exonuclease activity) (Lodish et al., 1995). The ac-
cessible residues property showed the best character-
ization, after molecular weight, for nuclease activity
function.
Disease-resistance genes are important in the cells
for the detection of pathogens and induction of de-
fense responses (Bai et al., 2002). These genes
code for proteins that interact selectively and non-
covalently with a nucleotide or any compound by
nucleotide binding sites (NBS). The NBS can affect
the disease resistance (R) protein function through
nucleotide binding (NtBind) or hydrolysis (Martin
et al., 2003). As shown in coiled coil, parallel
β − strand, total β − strand and α helix are the best
amino acid properties that represent this NtBind func-
tion. This can be explained by the fact that some
proteins contain a coiled coil domain, and the struc-
tural conformation of the NBS domain according to
the SCOP classification are α and β subunits (Wilson
et al., 2009).
Proteins with sequence-specific DNA binding
transcription factor activity (TranscFact) func-
tion interacts selectively and non-covalently with a
specific DNA sequence in order to modulate the
transcription of genetic information from DNA to
mRNA (Barrell et al., 2009). The amino acid com-
position and molecular weight are the properties that
best represented this function. The TranscFact class
is the function with the highest number of preserved
motifs (Figure 3). Two conserved prototype motifs
are analyzed using the web tool ScanProsite. Prosite
consists of documentation entries describing protein
domains, families and functional sites (Gattiker et al.,
2002).
The prototype motif 1 belongs to the WRKY do-
main that is an amino acid region defined by the con-
PredictingMolecularFunctionsinPlantsusingWavelet-basedMotifs
143

Table 2: Sensitivity, Specificity and Geometric mean values over 9 funcional classes.
Function Blast2GO Wavelet Pepstats-SVM
S
n
S
p
Gm S
n
S
p
Gm S
n
S
p
Gm
NtBind 0.609 0.67 0.639 0.864 0.739 0.799 0.423 0.705 0.546
TranscFact 0.854 0.771 0.811 0.756 0.731 0.744 0.619 0.837 0.72
RnaBind 0.571 0.809 0.68 0.810 0.756 0.782 0.545 0.755 0.642
Nase 0.545 0.866 0.69 1.000 0.772 0.878 0.545 0.698 0.617
RecepBind 0.818 0.928 0.871 1.000 0.8624 0.929 0.636 0.9092 0.758
Transp 0.741 0.729 0.735 0.741 0.754 0.748 0.618 0.643 0.63
LipBind 0.3 0.886 0.515 0.900 0.794 0.845 0.455 0.688 0.559
Kinase 0.884 0.633 0.748 0.691 0.794 0.740 0.533 0.702 0.612
EnzReg 0.316 0.93 0.542 0.778 0.817 0.797 0.636 0.784 0.706
0.626 0.802 0.692 0.838 0.780 0.807 0.557 0.746 0.643
Figure 3: Logos of conserved prototype motifs for Transc-
Fact molecular function. The motifs 1 and 2 correspond to
plant transcription factors WRKY and AP2/ERF domains,
respectively.
served amino acid sequence WRKYGQK and binds to
a specifically DNA sequence motif. The prototype
motif 2 is found in the AP2/ERF domain. The struc-
ture of this domain integrates a three-stranded β-sheet
and several α helices almost parallel to the β-sheets.
It contacts DNA via Arg and Trp residues located in
the β-sheet (Gattiker et al., 2002).
Having analyzed the prediction performances, an
ensemble of classifiers was trained with the best fea-
tures for each class. Those features are marked with
circles in Figure 2.
By comparing the achieved results shown in Ta-
ble 2, where the highest performances per class are
highlighted in bold, it is possible to infer that the pro-
posed wavelet-based method outperforms the other
methods in seven out of nine classes. The classifi-
cation results of the proposed method are lower than
the results of
Blast2GO
in only two cases, namely
TransFact and Kinase. Moreover, it can be seen that
the proposed method is the most sensitive of the three
methods shown, decreasing the achieved number of
false negatives. Geometric mean between sensitivity
and specificity is computed as a global performance
measure, showing that the wavelet based method-
ology overpasses the performance of
Blast2GO
on
about 11.5% in average and
Pepstats-SVM
in a
16.4%.
4 CONCLUSIONS
In this paper a methodology to molecular function
prediction in plants is proposed. The approach ex-
plores the distribution of the proteins computing a
set of prototype motifs. Thus, this motifs are used
to train a classifier an make a prediction to improve
the performance of the two novel explored methods
Pepstats
and
Blast2GO
. For this purpose an en-
hanced version of the previous work (Arango-Argoty
et al., 2011) was used, whose main feature is the
use of the continuous wavelet transform to identify
and characterize protein motifs. This transform can
provide accurate information about the structure of
a protein and hence the structures/motifs related to
each molecular function. Due to the protein database
contains sequences with a low identity (< 40%), the
prototype motifs showed to be discriminative and
representative. Thus, the classification performance
based on wavelet-motif detection improves the results
achieved by 1) a method based on global features of
the proteins (Pepstats), showing that a simple peptide
statistics are not enough to classify GO terms and 2) a
method based on similituds (Blast2GO) due to it ap-
proach lose sensitivity when the identity among se-
quences is low. At last, the proposed methodology
offers a more complete interpretation of the obtained
results since: a) the method is able to distinguish the
most representative properties of the amino acids for
each class and b) it identifies the motifs associated
with each molecular function. A possible direction
of research could be the use of robust methods for
clustering and computation of prototype motif such
as hidden Markov models.
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
144

ACKNOWLEDGEMENTS
This work was partially funded by the Research
office (DIMA) at the Universidad Nacional de
Colombia at Manizales and the Colombian National
Research Centre (COLCIENCIAS) through grant
No.111952128388 and the “Jovenes Investigadores e
Innovadores 2010”, Convenio Interadministrativo Es-
pecial de Cooperacion No. 146 de enero 24 de 2011
between COLCIENCIAS and Universidad Nacional
de Colombia Sede Manizales
REFERENCES
Arango-Argoty, G., Jaramillo-Garz´on, J. A., R¨othlisberger,
S., and Castellanos-Dom´ınguez, C. G. (2011). Pro-
tein subcellular location prediction based on variable-
length motifs detection and dissimilarity based classi-
fication. Annual International Conference of the IEEE
EMBS, (76).
Bai, J., Pennill, L., Ning, J., Lee, S., Ramalingam, J.,
Webb, C., Zhao, B., Sun, Q., Nelson, J., Leach, J.,
et al. (2002). Diversity in nucleotide binding site–
leucine-rich repeat genes in cereals. Genome research,
12(12):1871.
Barrell, D., Dimmer, E., Huntley, R., Binns, D.,
O’Donovan, C., and Apweiler, R. (2009). The GOA
database in 2009–an integrated Gene Ontology Anno-
tation resource. Nucleic acids research, 37(Database
issue):D396.
Chawla, N., Bowyer, K., Hall, L., and Kegelmeyer, W.
(2002). SMOTE: synthetic minority over-sampling
technique. Journal of Artificial Intelligence Research,
16(1):321–357.
Cheng, B., Carbonell, J., and Klein-Seetharaman, J. (2005).
Protein classification based on text document classifi-
cation techniques. Proteins: Structures, Function and
Bioinformatics, 58:955–970.
Conesa, A. and G¨otz, S. (2008). Blast2GO: A Compre-
hensive Suite for Functional Analysis in Plant Ge-
nomics. International journal of plant genomics,
2008:619832.
Gattiker, A., Gasteiger, E., and Bairoch, A. (2002). Scan-
Prosite: a reference implementation of a PROSITE
scanning tool. Applied Bioinformatics, 1(2):107–108.
Gupta, R., Mittal, A., Singh, K., Narang, V., and Roy, S.
(2009). Time-series approach to protein classification
problem. Engineering in Medicine and Biology Mag-
azine, 28(4):32–37.
Huang, Y., Niu, B., Gao, Y., Fu, L., and Li, W. (2010). Cd-
hit suite: a web server for clustering and comparing
biological sequences. Bioinformatics, 26(5):680–682.
Jain, E., Bairoch, A., Duvaud, S., Phan, I., Redaschi, N.,
Suzek, B., Martin, M., McGarvey, P., and Gasteiger,
E. (2009). Infrastructure for the life sciences: de-
sign and implementation of the UniProt website. BMC
bioinformatics, 10(1):136.
Johnson, M., Zaretskaya, I., Raytselis, Y., Merezhuk, Y.,
McGinnis, S., and Madden, T. (2008). Ncbi blast: a
better web interface. Nucleic acids research, 36(suppl
2):W5–W9.
Kawashima, S. and Kanehisa, M. (2000). Aaindex:
amino acid index database. Nucleic acids research,
28(1):374.
Lin, H., Han, L., Zhang, H., Zheng, C., Xie, B., and Chen,
Y. (2006). Prediction of the functional class of lipid
binding proteins from sequence-derived properties ir-
respective of sequence similarity. Journal of lipid re-
search, 47(4):824.
Liu, X., Korde, N., Jakob, U., and Leichert, L. (2006).
CoSMoS: conserved sequence motif search in the pro-
teome. BMC bioinformatics, 7(1):37.
Lodish, H., Berk, A., Zipursky, S., Matsudaira, P., Balti-
more, D., and Darnell, J. (1995). Molecular cell biol-
ogy. New York.
Martin, G., Bogdanove, A., and Sessa, G. (2003). Under-
standing the functions of plant disease resistance pro-
teins. Annual review of plant biology, 54(1):23–61.
Murray, K., Gorse, D., and Thornton, J. (2002). Wavelet
transforms for the characterization and detection of
repeating motifs1. Journal of molecular biology,
316(2):341–363.
Sarac¸, O. (2010). GOPred: GO Molecular Function Predic-
tion by Combined Classifiers. PloS one, 5(8):1–11.
Schneider, T. (2002). Consensus sequence zen. Applied
bioinformatics, 1(3):111.
Shen, Y. and Burger, G. (2010). TESTLoc: protein sub-
cellular localization prediction from EST data. BMC
bioinformatics, 11(1):563.
Swarbreck, D., Wilks, C., Lamesch, P., Berardini, T. Z.,
Garcia-Hernandez, M., Foerster, H., Li, D., Meyer,
T., Muller, R., Ploetz, L., Radenbaugh, A., Singh,
S., Swing, V., Tissier, C., Zhang, P., and Huala, E.
(2008). The arabidopsis information resource (tair):
gene structure and function annotation. Nucleic acids
research, 36.
Vinga, S. and Almeida, J. (2003). Alignment-free sequence
comparison: a review. Bioinformatics, 19(4):513.
Wheeler, D. (2002). Selecting the right protein-scoring ma-
trix. Current Protocols in Bioinformatics, pages 3–5.
Wilson, D., Pethica, R., Zhou, Y., Talbot, C., Vogel, C.,
Madera, M., Chothia, C., and Gough, J. (2009).
Superfamilysophisticated comparative genomics, data
mining, visualization and phylogeny. Nucleic acids
research, 37(suppl 1):D380.
Yu, L. and Liu, H. (2003). Feature selection for high-
dimensional data: A fast correlation-based filter so-
lution. In Machine Learning-International Workshop
then Conference-, volume 20, page 856.
PredictingMolecularFunctionsinPlantsusingWavelet-basedMotifs
145
