
Equidistant Reorder Operator for Cartesian Genetic Programming
Henning Cui
1 a
, Andreas Margraf
2 b
and J
¨
org H
¨
ahner
1 c
1
Institute for Computer Science, University of Augsburg, Am Technologiezentrum 8, 86159 Augsburg, Germany
2
Fraunhofer IGCV, Am Technologiezentrum 2, 86159 Augsburg, Germany
Keywords:
Cartesian Genetic Programming, CGP, Mutation Operator, Reorder, Genetic Programming, Evolutionary
Algorithm.
Abstract:
The Reorder operator, an extension to Cartesian Genetic Programming (CGP), eliminates limitations of the
classic CGP algorithm by shuffling the genome. One of those limitations is the positional bias, a phenomenon
in which mostly genes at the start of the genome contribute to an output, while genes at the end rarely do.
This can lead to worse fitness or more training iterations needed to find a solution. To combat this problem,
the existing Reorder operator shuffles the genome without changing its phenotypical encoding. However, we
argue that Reorder may not fully eliminate the positional bias but only weaken its effects. By introducing a
novel operator we name Equidistant-Reorder, we try to fully avoid the positional bias. Instead of shuffling the
genome, active nodes are reordered equidistantly in the genome. Via this operator, we can show empirically
on four Boolean benchmarks that the number of iterations needed until a solution is found decreases; and
fewer nodes are needed to efficiently find a solution, which potentially saves CPU time with each iteration. At
last, we visually analyse the distribution of active nodes in the genomes. A potential decrease of the negative
effects of the positional bias can be derived with our extension.
1 INTRODUCTION
Cartesian Genetic Programming (CGP) is a form
of Genetic Programming (GP), based on directed
acyclic graphs whose nodes are arranged in a two-
dimensional grid.
Since its inception in 1999 by Miller, CGP has
been used for various applications (Miller, 1999).
Originally, Miller used it to evolve digital cir-
cuits (Miller, 1999), which is still a use case to this
day (Froehlich and Drechsler, 2022; Manazir and
Raza, 2022). Furthermore, the CGP concept is used
in a diverse field of problem domains like classifica-
tion or regression (Miller, 2011). Other works uti-
lized CGP for image segmentation (Leitner et al.,
2012; Margraf et al., 2017) or the evaluation of sensor
data (Huang et al., 2022; Bentley and Lim, 2017).
CGP has some advantages over GP, such as the
absence of bloat (Miller, 1999): Solutions becom-
ing larger without an increase in fitness. However,
there are also some downsides and limitations in CGP.
One of those limitations is its positional bias, an ef-
a
https://orcid.org/0000-0001-5483-5079
b
https://orcid.org/0000-0002-2144-0262
c
https://orcid.org/0000-0003-0107-264X
fect which potentially limits CGP to fully explore
its search space (Goldman and Punch, 2013a). To
counteract this problem, Goldman and Punch intro-
duced Reorder, an operator to shuffle CGPs genome
ordering without changing its phenotype (Goldman
and Punch, 2013a). However, we believe that Re-
order suffers from some limitations, too, which could
lead to the search space being explored inefficiently.
This, in turn, could increase the iterations needed for
training as CGP gets stuck in local optima. To miti-
gate this problem, we introduce a modification to Re-
order: Equidistant-Reorder. With our modification,
the genome is not shuffled randomly anymore. By
additionally influencing the genomes reordering, the
effects of the positional bias may be further lessened.
In turn, CGP with Equidistant-Reorder needs less it-
erations to find a solution, which can save computa-
tional time.
In the following Section 2, we provide a quick
overview of the core principles of CGP and reintro-
duce the Reorder operator. Afterwards, Section 3
gives an overview of related work. In Section 4,
we discuss the aforementioned limitation of Reorder
more in-depth. Furthermore, we formally introduce
and explain our operator: Equidistant-Reorder. Af-
64
Cui, H., Margraf, A. and Hähner, J.
Equidistant Reorder Operator for Cartesian Genetic Programming.
DOI: 10.5220/0012174100003595
In Proceedings of the 15th International Joint Conference on Computational Intelligence (IJCCI 2023), pages 64-74
ISBN: 978-989-758-674-3; ISSN: 2184-3236
Copyright © 2023 by SCITEPRESS – Science and Technology Publications, Lda. Under CC license (CC BY-NC-ND 4.0)

terwards, its performance is analysed using several
Boolean benchmark problems in Section 5. At last,
Section 6 summarizes our findings and discusses fu-
ture research directions.
2 CARTESIAN GENETIC
PROGRAMMING
In the following section, the core principles of the su-
pervised learning algorithm called Cartesian Genetic
Programming (CGP) is reintroduced. In addition, the
CGP’s extension Reorder by Goldman and Punch is
presented (Goldman and Punch, 2013a).
The standard CGP variant described in Section 2.1
will be called STANDARD in the next sections to im-
prove readability. Apart from that, a CGP variant with
the Reorder operator employed will be called RE-
ORDER.
2.1 Representation
Traditionally, CGP is represented as a directed,
acyclic and feed-forward graph. This graph contains
nodes which are arranged in a c × r grid, with c ∈ N
+
and r ∈ N
+
defining the number of columns and rows
in the grid respectively. Given an arbitrary amount
of program inputs, the CGP model feeds those inputs
forward through its partially connected nodes to get
any desired amount of outputs. Please note that, with
today’s standards, a CGP model typically consists of
only one row (Miller, 2011). That means, r = 1.
The set of nodes can be divided into three groups:
input-, computational- and output-nodes. Input nodes
directly receive a program input. Their only purpose
is to relay the program input. Differently, computa-
tional nodes are represented by a function gene and
a connection genes. Here, a ∈ N
+
is the maximum
arity of all functions in the predefined function set,
while the function gene addresses the encoded com-
putational function of the node. Please note that if a
function needs less than a inputs, all excessive input
genes are ignored. At last, output nodes redirect the
calculated output of a computational node and define
the model’s output. They are represented by a sin-
gle connection gene, which defines a path between a
previous node and the current node.
In this sense, when we mention a graph with
N ∈ N
+
nodes, this graph will have only one row, N
computational nodes and additional input and output
nodes corresponding to the given learning task.
Another important distinction of computational
nodes are active and inactive nodes. Active nodes are
part of a path to an output node. Hence, they actively
n
0
:
INPUT
n
1
:
INPUT
n
2
:
ADD
n
3
:
SUB
n
4
:
ADD
n
5
:
OUTPUT
Figure 1: Example graph defined by a CGP genotype. The
dashed node and connections are inactive due to not con-
tributing to the output.
contribute to a program output. On the contrary, inac-
tive nodes do not contain a path to an output node and
do not contribute to the model’s output. However, in-
active nodes are essential to CGP as they probably aid
the evolutionary process through genetic drift (Turner
and Miller, 2015).
An illustrative example of a graph defined by a
CGP genotype can be seen in Figure 1. It shows a
graph with c = 6 and r = 1, which is supposed to solve
a task consisting of two inputs and one output. Here,
the first two nodes (n
0
and n
1
) are input nodes and
provide two different program inputs. The follow-
ing nodes (n
2
–n
4
) are computational nodes. At last,
the output is provided by one output node n
5
. In this
example, both inputs are subtracted, with this inter-
mediate result being added to itself and taken as the
program output. Thus, every node drawn with a solid
line is an active node. The node n
2
is an inactive node
(marked by dashed lines) since it does not contribute
to the program output nor contains a path to an output
node.
2.2 Evolutionary Algorithm Used by
CGP
As is standard in most CGP variants, an elitist (µ + λ)
evolution strategy with µ = 1 and λ = 4 is used to
select the best graph. This is combined with neu-
tral search to improve performance and convergence
time (Turner and Miller, 2015; Yu and Miller, 2001;
Vassilev and Miller, 2000). Here, neutral search
means that, when an offspring and the current par-
ent have the same fitness value, the offspring is al-
ways chosen as the next parent. Hence, by always
preferring the child, neutral drift is allowed to oc-
cur. For that reason, the search algorithm is explor-
ing various possible solutions given by different geno-
types (Miller, 2020).
Regarding its mutation operator, a point mutation
is oftentimes found in literature (Miller, 2011; Miller,
2020). Here, the operator simply iterates over all
genes, mutating each with a probability defined by
the user (Miller, 2011). However, for this mutation
operator, it is possible that only inactive genes are mu-
tated. As no active node is changed, it is not possible
to evaluate the quality of the newly mutated program.
This could potentially lead to more training iterations
Equidistant Reorder Operator for Cartesian Genetic Programming
65

needed and introduce fitness plateaus.
In order to prevent such plateaus, Goldman and
Punch propose an alternative mutation operator which
they call Single (Goldman and Punch, 2013b). For
this method, randomly selected genes are mutated un-
til a gene corresponding to an active node is mutated.
This approach has the advantage that no wasted eval-
uations are performed and a change in the phenotype
is guaranteed. Furthermore, as inactive nodes are mu-
tated too, genetic drift is also able to occur. Thus,
when the optimal mutation rate is not known, the au-
thors Goldman and Punch claim that Single should be
preferred (Goldman and Punch, 2013b).
2.3 Extension: Reorder
By enforcing a feed forward grid, Goldman and
Punch found a negative impact in CGPs search
space (Goldman and Punch, 2013a). One insight is
a positional bias: a non-uniformity of the probabil-
ity that a node is active. When nodes are closer to
the input nodes, their probability of becoming active
is higher. The other way round, nodes closer to the
output nodes have the lowest probability of becoming
active.
During the mutation of a connection gene, each
previous node has the same probability of being cho-
sen as the new starting node for the connection. How-
ever, nodes near input nodes have more options for
successor nodes, as directed edges are only allowed
from left to right. This is why they have a higher
chance of becoming active, as a node is only active
when it is part of a path to an output node. Nodes
closer to output nodes have less options of being cho-
sen by other nodes, as they have less nodes in front.
With the help of Figure 1, the positional bias can
be exemplified. In this example, n
1
’s possible suc-
cessors are n
2
–n
5
whereas n
4
’s possible successor is
just n
5
. Hence, four nodes can potentially mutate their
connection to n
1
and if one of those nodes is active, n
1
becomes active, too. Contrary, n
4
has only one node
in front which could mutate its connection to n
4
.
This leads to some drawbacks, as this positional
bias makes it difficult or impossible to solve certain
problems (Goldman and Punch, 2013a; Payne and
Stepney, 2009). To combat such limitations, Goldman
and Punch proposed two new operators which are ex-
ecuted before mutation: DAG and Reorder (Goldman
and Punch, 2013a). The first extension, DAG, allows
each node to mutate connections to every other node
in the genome as long as the new connection does not
create a cycle. However, as we do not build upon it,
we will not discuss DAG in further detail. As for Re-
order, this extension shuffles the genome’s node or-
dering, with the result of a shuffled genotype. Nev-
ertheless, by respecting the sequence of operations of
the active nodes, the phenotype does not change.
This Reorder operator begins by creating a depen-
dency set D, containing each computational node and
the nodes from which it gets its input from. In ad-
dition, the new genome is initialized. Here, the new
genome contains all input and output nodes from the
original genome, as they do not get assigned a new
position. Afterwards, the set of addable nodes Q is
created. Initially, this set contains only computational
nodes whose dependencies are satisfied. Let b be
a computational node, and a be a computational or
input node from which b gets one input from, with
(b, a) ∈ D. This means, there is a directed edge con-
necting a to b via b’s connection gene. Then, in this
context, b is satisfied when all nodes from which b
gets its input from are mapped into the new genome.
With both sets initialized, the shuffling of the
genome is done by repeating the following steps:
1. Select and remove a random node a ∈ Q.
2. Map a sequentially to the next, free location in the
new genome.
3. For each node pair (b, a) ∈ D with b depending on
a, do:
(a) If all dependencies of b are satisfied, add b to
Q.
This operator ends when all computational nodes
have been assigned a new location in the genotype.
By always satisfying each nodes dependency, the
phenotype stays the same. Hence, the fitness value of
the genome does not change by re-evaluating. How-
ever, by permutating the genome ordering, the authors
of (Goldman and Punch, 2013a) show a theoretical
improvement of a mutation. This improvement is also
shown empirically on four different benchmarks, as
utilizing Reorder can lead to fewer iterations needed
until a solution is found (Goldman and Punch, 2015;
Goldman and Punch, 2013a).
3 RELATED WORK
Our work focuses on improving the Reorder operator,
which has, to the best of our knowledge, not been ex-
plored yet. Nevertheless, various other articles layed
out the foundation for this work.
Goldman and Punch detected the aforementioned
positional bias (Goldman and Punch, 2013a; Gold-
man and Punch, 2015). To mitigate its effects, they
introduced Reorder and DAG.
Moreover, other works explore new operators for
CGP and / or its search space and its limitations, too.
ECTA 2023 - 15th International Conference on Evolutionary Computation Theory and Applications
66
There are various algorithms which extend the basic
CGP formula. One such extension was developed by
Kalkreuth (Kalkreuth, 2022). By introducing two new
operators to duplicate or swap genes in the phenotype,
CGP is able to find a solution in less iterations.
Another possibility of changing CGPs geno-
and phenotype was introduced by Walker and
Miller (Walker and Miller, 2004). With their ex-
tension, nodes or subgraphs in the genotype can be
dynamically created, evolved, reused and removed.
This leads to higher fitness values and less iterations
needed to find good solutions.
Harding et al. included a multitude of additional
functions in the function pool (Harding et al., 2011).
These functions directly change CGPs phenotype and
allows it to solve problems with extremely large num-
bers of inputs. Some of these additional functions
were then reused to allow CGP to solve image pro-
cessing tasks, as was done by Leitner et al. or Harding
et al. (Leitner et al., 2012; Harding et al., 2013).
At last, Wilson et al. (Wilson et al., 2018) intro-
duced a different representation of CGP based on a
floating point representation. In addition, they in-
cluded operators to change the genotype, too, which
enabled them to add or remove nodes or even whole
subgraphs.
CGPs search space is another important factor for
its performance. In the context of neural architecture
search, Suganuma et al. used a highly specified func-
tion set to reduce the size of CGPs search space (Sug-
anuma et al., 2020).
It is also possible to improve the search space by
using specific crossover operators, as was done by
Torabi et al. (Torabi et al., 2022).
4 EQUIDISTANT-REORDER
While REORDER has the potential to decrease the
number of iterations needed until a solution is found,
it is also possible that it does not fully reduce the
aforementioned limitation in the search space. This
Section argues a potential drawback of REORDER and
introduces CGP with Equidistant-Reorder, called E-
REORDER—a modified variant of REORDER to avoid
the flaw.
4.1 Potential Limitation of Reorder
With the Reorder operator, a node can only be as-
signed a new position in the grid when all its depen-
dencies are satisfied. This means that, at first, the only
satisfied node dependencies are nodes directly con-
nected to input nodes. Furthermore, computational
n
0
:
INPUT
n
1
:
FUNC.
n
2
:
FUNC.
n
3
:
FUNC.
n
5
:
OUTPUT
(a) A graph before Reorder. Input and output node are n
0
and n
5
, respectively. Nodes n
1
–n
3
are computational nodes,
with FUNC. being an arbitrary function.
n
0
:
INPUT
n
1
:
FUNC.
n
3
:
FUNC.
n
2
:
FUNC.
n
5
:
OUTPUT
(b) The graph after a potential Reorder.
Figure 2: A graph defined by a CGP genome, before and
after Reorder.
nodes near input nodes have a higher probability of
being connected to input nodes. The reason is that
these nodes have little or no computation nodes prior
to them. Hence, they are more prone to being con-
nected to input nodes compared to nodes near output
nodes.
As a consequence, computational nodes near the
input nodes have a higher probability of getting added
to the set of addable nodes earlier, compared to other
computational nodes. This leads to them having a
higher probability of being added earlier into to the
new, shuffled genome, too. Hence, they get assigned
a new position which is, again, closer to the input
nodes.
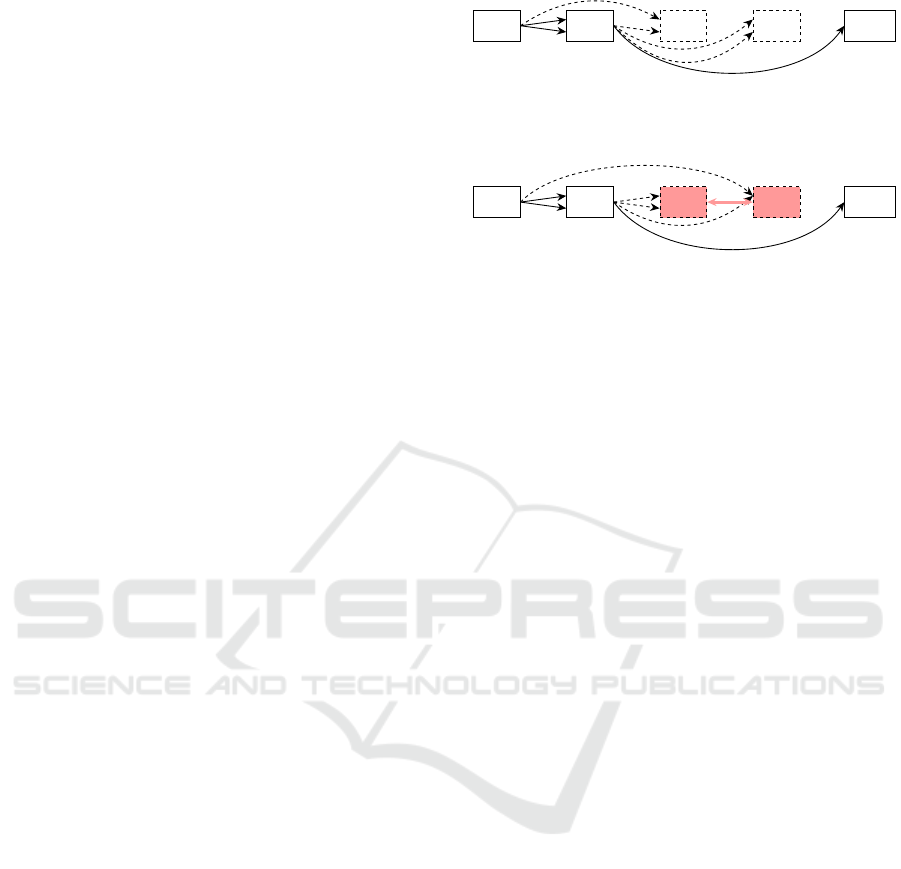
This limitation can be visualized by Figure 2.
Here, the aforementioned problem is illustrated. Fig-
ure 2a shows a graph before its reordering. As n
1
is
only connected to the input node, it is the only node
with satisfied dependencies. Hence, it is the only node
added to the addable set and will be reordered to the
first position in the new genome. Therefore, the only
active node of the graph will not change its position in
the grid. Generally, it is not possible for n
1
to change
its position in this graph setup with the Reorder oper-
ator.
After adding n
1
into the new genome, n
2
and n
3
have satisfied dependencies. They will be placed into
the genome afterwards and a potential solution can be
seen in Figure 2b.
4.2 Equidistant-Reorder Operator
To circumvent the limitation described in Section 4.1,
we propose a different strategy to reorder nodes. For
our method, all active nodes are placed equidistantly
apart in the grid first; hence we name our method
Equidistant-Reorder, and a CGP variant with our
method will be called E-REORDER in the remain-
der of this work. Furthermore, as mentioned in Sec-
Equidistant Reorder Operator for Cartesian Genetic Programming
67

tion 2.3, STANDARD suffers from positional bias. By
spreading active nodes over the whole genome, this
drawback should be lessened, too.
To perform E-REORDER on a genome with
N computational nodes, two sets must be ob-
tained. The first set A defined as A
:
= {a
1
, ·· · , a
n
|
a
i
is active computational node} contains all active
computational nodes. In addition, the set of
all inactive computational nodes
˜
A
:
= { ˜a
1
, ·· · , ˜a
m
|
˜a
i
is inactive computational node} is needed as well.
Afterwards, a new modified genome G is initialized.
Initially, G contains all input and output nodes from
the original genome, as these two node sets will not be
assigned a new position. Please note that the positions
of all input and output nodes are not changed in this
algorithm. Then, let s be the starting index and e be
the last index of computational nodes in the genome.
With these values, a set of equidistantly spaced
numbers L
:
=
bs + i ·
e−s
n
c | i = 1, ·· · , n
over the in-
terval [s, e] can be generated. Here, L is the set of
new positions for active nodes. To further illustrate
the generation of L, Algorithm 1 describes it more in-
depth. However, if n = 1, then L
:
= {e}. This means,
if there is only one active node, it will be placed at the
end of the genome just before the output nodes. As
a result, the active node is able to mutate its connec-
tion to an arbitrary computational node, which should
further lessen the positional bias.
On the contrary,
˜
L is the set of all genome loca-
tions without the positions of active nodes, which is
defined as
˜
L
:
= {s, s + 1, ··· , e} ∩ L
:
= {
˜
l
1
, ·· · ,
˜
l
N−n
}
with
˜
l
1
<
˜
l
2
< ··· <
˜
l
n
. Hence,
˜
L is the set of new
positions for inactive nodes.
Now, active computational nodes can be placed
into a new position in G by assigning a
i
∈ A the posi-
tion l
i
∈ L, for i = 1, ··· , n. As the order of computa-
tional nodes are not modified, the reordering does not
change the semantic and no re-evaluation is required.
Next, inactive nodes are placed in order into G by
assigning them into the next genome location without
an active node in G. As such, the new position of
an inactive node ˜a
j
∈
˜
A is defined by
˜
l
j
∈
˜
L with j =
1, ·· · , N − n.
At last, all connection genes must be corrected by
changing their connection from the old genome loca-
tion to the new one. Problems can arise in the case
of inactive nodes, as it is possible that a connection
gene gets assigned a new position in front of the cur-
rent node. In this case, a new connection to a previous
node must be mutated.
To better illustrate the algorithm of Equidistant-
Reorder, its workings are depicted in Algorithm 2.
Furthermore, Figure 3 shows two example graphs,
with Figure 3a depicting an example graph. Figure 3b
Function Lin-Space:
Data:
• start point s
• end point e
• number of evenly spaced values n
step ←
e − s
n
;
S ←
/
0;
i ← 1;
while i ≤ n do
S ← S ∪ bs + i · stepc;
i ← i + 1;
end
Algorithm 1: Generate a set of equidistantly spaced
numbers.
Function Equidistant-Reorder:
A ← all active computational nodes;
˜
A ← all inactive computational nodes;
initialize new genome G with the same
input and output nodes from the old
genome;
s ← |input nodes|;
e ← |input nodes| + |A| + |
˜
A|;
L ← Lin-Space(s, e,|A|);
˜
L ← {s, s + 1, ··· , e} ∩ L;
foreach (a, l) ∈ (A, L) do
G[l] ← a;
end
foreach
˜a,
˜
l
∈ (
˜
A,
˜
L) do
G
˜
l
← ˜a;
end
foreach node ∈ G do
foreach connection
i
belonging to
node do
update(connection
i
);
if connection
i
points to the front
then
mutate connection
i
;
end
end
end
return G;
Algorithm 2: Equidistant-Reorder function.
displays the graph after E-Reorder. Both active nodes
are placed evenly spaced into the new genome, with
the inactive node being assigned to the free position.
ECTA 2023 - 15th International Conference on Evolutionary Computation Theory and Applications
68

n
0
:
INPUT1
n
1
:
INPUT2
n
2
:
FUNC.
n
3
:
FUNC.
n
4
:
FUNC.
n
5
:
FUNC.
n
6
:
FUNC.
n
7
:
FUNC.
n
8
:
OUTPUT
(a) A graph before E-Reorder. Input and output node are n
0
, n
1
and n
8
. Nodes n
2
–n
7
are computational nodes, with FUNC.
being an arbitrary function.
n
0
:
INPUT1
n
1
:
INPUT2
n
2
:
FUNC.
n
3
:
FUNC.
n
5
:
FUNC.
n
4
:
FUNC.
n
7
:
FUNC.
n
6
:
FUNC.
n
8
:
OUTPUT
(b) The graph after a potential E-Reorder, according to Algorithm 2. Node n
7
must mutate new connections to prevent
connections backwards from the new position of n
6
to n
7
.
Figure 3: A graph defined by a CGP genome, before and after E-Reorder.
4.3 Time Complexity
The modifications of Equidistant-Reorder compared
to the original Reorder operator from Goldman and
Punch have the same additional runtime complex-
ity (Goldman and Punch, 2013a). In the following,
let N ∈ N
+
be the number of computational nodes of
the considered CGP graph.
To obtain the set of all active and inactive compu-
tational nodes, O (a ·N) time is needed, where a ∈ N
+
is the arity of the nodes. Other sets, like L or
˜
L, need
O(N) time. The placing of active and inactive nodes
into a new genome takes only O(N) time, as this only
requires iterating through the sets of active and inac-
tive nodes and their respective new positions once. At
last, the connection genes have to be updated. Never-
theless, this process takes only O(c · N) time by iter-
ating through every computational node and updating
its c connection genes. Please note, a ≡ c, as the high-
est arity a is also the number of connection genes c.
Thus, Equidistant-Reorder needs O(a · N) time,
which is the same as Reorder (Goldman and Punch,
2015).
5 EVALUATION OF E-REORDER
In order to find out whether the modification we pro-
posed for Reorder truly benefits CGP, we conducted
an empirical study
1
. This section describes our exper-
1
Implementation and benchmarks can be found at https:
//github.com/CuiHen/Equidistant Reorder.
imental design. Afterwards, we attempt to answer the
following two questions:
Q1 Which model needs, considering the learning
task, less training iterations: REORDER or E-
REORDER?
Q2 As mentioned in Section 4.1, REORDER may not
reduce the limitation in the search space of CGP.
How does E-REORDER perform compared to RE-
ORDER?
5.1 Experimental Design
In this section, the evaluated CGP configurations and
utilized benchmarks are presented. Afterwards, we
briefly describe the Bayesian models used to evaluate
and rank different CGP configurations. With these
models, the performance of multiple configurations
can be compared by calculating a probability of each
configuration being the best.
5.1.1 Settings of CGP
For our experiments, we compared each combination
of STANDARD, REORDER and E-REORDER in con-
junction with two different mutation strategies: point
mutation and Single. Regarding the hyperparameters,
when the mutation operator Single is used, the only
hyperparameter is the number of nodes N. Thus, we
investigated N ∈ {50, 100, 150, ·· · , 2000} and each N
was tested 75 times with independent repetitions and
different random seeds.
As for the point mutation, the number of nodes N
and the mutation probability p must be determined.
Equidistant Reorder Operator for Cartesian Genetic Programming
69

For this task, we performed a hyperparameter search
using a Tree-structured Parzen Estimator
2
. The algo-
rithm performed a total of 100 trials to find good hy-
perparameters. For each trial, the configuration was
tested 10 times with independent repetitions and dif-
ferent random seeds. These results were then aver-
aged to obtain a single value. Runs were terminated
when a model is able to solve a benchmark com-
pletely.
Considering the evolutionary algorithms, we uti-
lized a standard (1+4)-ES as described in Section 2.2
for all trainings. Furthermore, to be comparable to
the original evaluations by Goldman and Punch, we
also compared different mutation strategies as already
mentioned above (Goldman and Punch, 2015; Gold-
man and Punch, 2013a). The original works com-
pared two different strategies for the point mutation:
skip and accumulate. However, they did not change
the behaviour of the point mutation but save compu-
tational time only. Furthermore, the authors of the
work (Goldman and Punch, 2015) found no signifi-
cant difference between those two operators. Because
of that, we employed their point mutation strategy of
skip. Offsprings, whose phenotype are equal to the
parents phenotype, are not evaluated. Instead, they
get their parents fitness value assigned.
5.1.2 Problem Set
To evaluate the different settings, we used four
Boolean benchmark problems: 3-bit Parity, 16-4-bit
Encode, 4-16-bit Decode and 3-bit Multiply. In the
following, we will call these Parity, Encode, De-
code and Multiply, respectively. Parity is regarded
as too easy by the Genetic Programming commu-
nity (White et al., 2013). Nevertheless, it is com-
monly used as a benchmark in the literature (Yu
and Miller, 2001; Kaufmann and Kalkreuth, 2020;
Kaufmann and Kalkreuth, 2017). Hence, we also in-
cluded it in our evaluations for ease of comparison.
Encode and Decode are problems with different in-
put and output sizes. At last, Multiply is a compar-
atively hard problem (Walker and Miller, 2008), and
recommended by White et al. (White et al., 2013).
All four benchmarks are used to evaluate REORDER,
too, which is also why we utilized them (Goldman
and Punch, 2013a; Goldman and Punch, 2015).
As we employed four Boolean benchmark prob-
lems, we also trained them with the standard function
set for these problems. They contain the Boolean op-
erators AND, OR, NAND and NOR.
These benchmarks also lead to a standard fitness
2
For the hyperparameter search, we utilized the Python
library Optuna (Akiba et al., 2019).
function, which is defined by the ratio of correctly
mapped inputs. Let f : {0,1}
n
→ {0, 1}
m
be a correct
Boolean mapping for n ∈ N
+
inputs and m ∈ N
+
out-
puts. Then, the fitness of an individual g : {0, 1}
n
→
{0, 1}
m
, which relates to the learning task f , is defined
as follows:
|{x ∈ {0, 1}
n
| f (x) = g(x)}|
|{0, 1}
n
|
(1)
5.1.3 Bayesian Data Analysis
In search of a good number of nodes for Single, mul-
tiple configurations were investigated. Furthermore,
to present our final results, six different, final settings
were compared. In all those cases, each configuration
has to be ranked to find the respective best solution.
However, we only considered the number of training
iterations, a unit which can not be negative. Thus,
other common distributions such as Student’s t distri-
butions can not be expected to model the data well.
Hence, we performed a Bayesian data analysis
3
for
the posterior distributions of our results. The model
to compare the algorithms is based on the Plackett-
Luce model described by Calvo et al. (Calvo et al.,
2018). It allows the computation of a set of ranked
options by estimating the probabilities of each of the
options to be the one with the highest rank.
In addition, in our final results, we report for
each configuration the 95 % highest posterior den-
sity intervals (HPDI) of the distribution of µ
con fig
,
where µ
con fig
is a random variable corresponding to
the respective mean numbers of iterations until solu-
tion. At that, the distribution of µ
con fig
is estimated
by the gamma distribution–based model for compar-
ing non-negative data from cmpbayes (P
¨
atzel, 2023).
Please note, a 95 % HPDI interval [l, u] can be read as
p(l ≤ µ
con fig
≤ u) = 95 %. This means, the probabil-
ity of the algorithms results lying between the bounds
l and u has a probability of 95 %.
On another note, prior sensitivity analyses were
conducted prior to ensure the robustness of all mod-
els. As they always display similar results, ro-
bust and meaningful models are implicated. Finally,
please note that cmpbayes uses Markov Chain Monte-
Carlo sampling to obtain its distributions. There-
fore, the usual checks to ensure convergence and well-
behavedness (trace plots, posterior predictive checks,
ˆ
R values, effective sample size) were performed. For
more information regarding the models, we refer to
Kruschke and P
¨
atzel (Kruschke, 2013; P
¨
atzel, 2023).
3
We utilized the Python library cmpbayes (P
¨
atzel, 2023)
for all statistical models.
ECTA 2023 - 15th International Conference on Evolutionary Computation Theory and Applications
70

5.2 Performance on Training Iterations
As already mentioned, we performed a hyperparame-
ter search and determined the best configuration via a
Plackett-Luce model described by Calvo et al. (Calvo
et al., 2018).
The best results found by the hyperparameter
search can be seen in Table 1. For each benchmark,
the configurations are sorted from best to worst in de-
scending order. We report the CGP variant and its
mutation operator. Furthermore, we list, with their re-
spective averaged number of iterations needed until
a solution is found, its HPDI, the number of nodes
needed, their mutation rate, the number of active
nodes and the probability of a solution being the best.
For all four benchmarks, our novel variant E-
REORDER probably performed best. For Encode and
Decode, E-REORDER with Single lead to the best re-
sults while Encode and Parity performs best with the
point mutation. This could also mean that we were
not able to find the best mutation rate with our hy-
perparameter search. Albeit when looking at the re-
sults for E-REORDER in the context of Encode, the
differences of both mutation operators are almost neg-
ligible. The average number of iterations needed are
almost identical and their HPDI are close, too. Al-
though, according to the Calvo model, the probabili-
ties for E-REORDER with its respective mutation op-
erators are not equal but differ about 5 percentage
points—indicating a preference for the point muta-
tion. On another note, Single needs less nodes to find
a solution compared to the point mutation.
Generally, E-REORDER with Single tends to need
the least amount of computational nodes. While
STANDARD with point mutation needs the least
amount of nodes for Multiply, it is also the worst al-
gorithm in this case. This may also be a reason why
E-REORDER needs the least amount of iterations until
a solution is found. With less nodes, the search space
for E-REORDER decreases, too. Hence, it may find
a solution faster. Furthermore, with less nodes, CPU
time can be saved with each iteration, as less potential
computations take place.
Interestingly, E-REORDER and REORDER with
the point mutation tend to have similar mutation rates.
Except for Encode, both rates only differ marginally
and varies vastly from STANDARD’s mutation rate. In
our experiments, STANDARD favours a lower muta-
tion rate compared to the other two algorithms, which
could also indicate that REORDER and E-REORDER
favours a higher change in the genotype per mutation.
Another factor for E-REORDER’s better perfor-
mance may be the additional mutations of inactive
nodes. With their reordering, some connection genes
0 20 40 60 80 100
Relative node index in % to the graph's total length
0.0
0.2
0.4
0.6
0.8
1.0
% active during training
Multiply
E-Reorder
Reorder
Standard
Figure 4: Active node distribution of STANDARD, RE-
ORDER and E-REORDER. Each distribution shows the av-
eraged result of 75 aggregated solutions for Multiply.
must be mutated to prevent backward-connections.
This could lead to more genetic drift, which may im-
prove its performance.
5.3 Distribution of Active Nodes
In Section 4.1, we hypothesize a potential limitation
of REORDER and speculate in Section 4.2 that E-
REORDER may further lessen the effects of a posi-
tional bias. Hence, one question we would like to an-
swer is the effect of E-REORDER on the distribution
of active nodes, compared to REORDER and STAN-
DARD. For this task, a visualization of the active node
distribution of the final results for all three CGP vari-
ants is given in Figure 4. The x-axis shows the rela-
tive node index in percent, while the y-axis shows the
probability of that node being active.
Please note that we only visualize our findings for
Multiply, as the other three benchmarks show almost
identical plots and behaviours. In addition, we always
compare the best configurations found, as reported in
Table 1. Again, we averaged the active node distribu-
tion of 75 independent repetitions with random seeds.
Furthermore, the figure only depicts the Single muta-
tion strategy, as this was the best mutation operator
for all three CGP variants. Besides, the visualization
for the point mutation strategy shows only marginal
differences compared to Single. At last, as the CGP
variants have different number of nodes, we do not re-
port the actual node indices in Figure 4. Instead, we
report the relative node index in percent to the graphs
total length.
In the context of STANDARD, the positional bias
can be clearly seen. At the beginning of the genome,
nodes have a high probability of being active. How-
ever, after the first 30 % of the genome, this probabil-
ity decreases to 10 % or less.
As for REORDER, this variant is able to moder-
ately overcome the bias. While it also contains a drop
in node activity, the decrease is far less pronounced
compared to STANDARD. Furthermore, the probabil-
ity of active nodes increases in the last 10 % of node
indices.
Equidistant Reorder Operator for Cartesian Genetic Programming
71

Table 1: Best results found for each E-REORDER, REORDER and STANDARD configuration. Algorithms are sorted from best
to worst in descending order. Here, Avg is the average number of iterations until a solution is found; # Nodes is the number of
computational nodes used; p(mut) is the mutation rate; # active is the number of active nodes for this solution; and p(best)
the probability of the solution being the best among the other six.
Variant Mutation Avg HPDI # Nodes p(mut.) # Active p(best)
Parity
E-REORDER Prob. 322 [278, 371] 350 0.081 129 0.26
STANDARD Single 342 [289, 404] 600 — 42 0.23
REORDER Prob. 400 [341, 471] 450 0.085 85 0.21
E-REORDER Single 471 [395,561] 50 — 27 0.16
REORDER Single 739 [606,902] 200 — 57 0.11
STANDARD Prob. 2,524 [2,063, 3,084] 750 0.009 43 0.02
Encode
E-REORDER Prob. 5,294 [4,656, 6,049] 650 0.014 336 0.32
E-REORDER Single 5,296 [4,759,5,908] 150 — 87 0.27
STANDARD Single 6,213 [5,550, 6,947] 200 — 65 0.22
REORDER Prob. 8,642 [7,644, 9,787] 550 0.018 249 0.12
REORDER Single 14,733 [12,786, 16,977] 600 — 258 0.04
STANDARD Prob. 20,378 [17,592, 23,557] 700 0.009 103 0.03
Decode
E-REORDER Single 12,119 [11,174, 13,142] 250 — 145 0.39
E-REORDER Prob. 14,596 [13,154, 16,153] 650 0.008 345 0.26
REORDER Single 15,017 [13,826, 16,295] 700 — 186 0.21
STANDARD Single 23,132 [20,959, 25,482] 400 — 242 0.07
STANDARD Prob. 28,510 [25,868, 31,349] 700 0.009 183 0.04
REORDER Prob. 32,902 [30,227, 35,857] 800 0.003 402 0.03
Multiply
E-REORDER Single 70,304 [59,647, 82,144] 700 — 362 0.41
REORDER Single 90,370 [79,273, 103,286] 750 — 118 0.23
STANDARD Single 129,739 [110,948, 150,742] 700 — 411 0.14
REORDER Prob. 119,426 [104,665, 135,512] 1000 0.013 540 0.13
E-REORDER Prob. 194,044 [163,843, 227,740] 700 0.012 363 0.08
STANDARD Prob. 414,639 [360,719, 472,413] 500 0.007 102 0.02
The distribution for E-REORDER differs com-
pared to the last two CGP variants. For the first 5 % of
nodes of graph, the probability for nodes being active
oscillates in the range of [0.2, 1.0]. Afterwards, it fluc-
tuates around a probability of 0.75 % with occasional
spikes in probability. Nevertheless, such fluctuat-
ing behaviour should be anticipated, as E-REORDER
places active nodes equidistantly apart. In addition,
by forcing a uniform distribution of active nodes in
combination with a better fitness value as seen in Sec-
tion 5.2, we believe that we lessened the effects of a
positional bias.
6 CONCLUSION
The existing Reorder operator introduced by Gold-
man and Punch is able to moderately overcome a po-
sitional bias, from which Cartesian Genetic Program-
ming (CGP) suffers (Goldman and Punch, 2013a).
Nonetheless, we theorized that Reorder might have
a small limitation, which may hinder a complete ex-
ploration of the search space. A node may only be
reordered into the new genome when all of its de-
pendencies are satisfied—which means that all in-
put nodes must be mapped into the genome before-
hand. This may slightly favour nodes near input
nodes, which could potentially limit Reorders full po-
tential.
ECTA 2023 - 15th International Conference on Evolutionary Computation Theory and Applications
72

With our novel modification to Reorder, called
Equidistant-Reorder, we are able to evade the limi-
tation of Reorder. The algorithm works by reordering
active nodes equidistantly apart throughout the whole
genome. As a result, CGP with Equidistant-Reorder
is able to find a solution for a given problem in less
iterations compared to the CGP baseline or with the
Reorder extension. In most cases, the total number of
nodes needed to train CGP is reduced, too.
As for future work, different reorder strategies
could be examined as we only focused on equidis-
tant spacing. It would also be possible to apply a
uniform distribution instead of enforcing an equidis-
tant distance. Another interesting aspect would be to
move all or the majority of active nodes to the end of
the genome. Then, there are almost no nodes with a
higher probability of becoming active. As all active
nodes are at the end of the genome, each node is able
to mutate a connection to an arbitrary node behind. It
may lead to less positional bias, too, but could also
lead to other potential problems.
ACKNOWLEDGEMENTS
The authors would like to thank the German Federal
Ministry of Education and Research (BMBF) for sup-
porting the project SaMoA within VIP+.
REFERENCES
Akiba, T., Sano, S., Yanase, T., Ohta, T., and Koyama, M.
(2019). Optuna: A next-generation hyperparameter
optimization framework. In Proceedings of the 25th
ACM SIGKDD International Conference on Knowl-
edge Discovery and Data Mining.
Bentley, P. J. and Lim, S. L. (2017). Fault tolerant fusion
of office sensor data using cartesian genetic program-
ming. In 2017 IEEE Symposium Series on Computa-
tional Intelligence (SSCI), pages 1–8. IEEE.
Calvo, B., Ceberio, J., and Lozano, J. A. (2018). Bayesian
inference for algorithm ranking analysis. In Proceed-
ings of the Genetic and Evolutionary Computation
Conference Companion, GECCO ’18, page 324–325,
New York, NY, USA. Association for Computing Ma-
chinery.
Froehlich, S. and Drechsler, R. (2022). Unlocking approxi-
mation for in-memory computing with cartesian ge-
netic programming and computer algebra for arith-
metic circuits. it - Information Technology, 64(3):99–
107.
Goldman, B. W. and Punch, W. F. (2013a). Length bias
and search limitations in cartesian genetic program-
ming. In Proceedings of the 15th Annual Conference
on Genetic and Evolutionary Computation, GECCO
’13, page 933–940, New York, NY, USA. Association
for Computing Machinery.
Goldman, B. W. and Punch, W. F. (2013b). Reducing
wasted evaluations in cartesian genetic programming.
In Genetic Programming, pages 61–72, Berlin, Hei-
delberg. Springer Berlin Heidelberg.
Goldman, B. W. and Punch, W. F. (2015). Analysis of
cartesian genetic programming’s evolutionary mech-
anisms. IEEE Transactions on Evolutionary Compu-
tation, 19(3):359–373.
Harding, S., Leitner, J., and Schmidhuber, J. (2013). Carte-
sian Genetic Programming for Image Processing,
pages 31–44. Springer New York, New York, NY.
Harding, S. L., Miller, J. F., and Banzhaf, W. (2011). Self-
Modifying Cartesian Genetic Programming, pages
101–124. Springer Berlin Heidelberg, Berlin, Heidel-
berg.
Huang, W., He, P., Yan, Z., and Wu, H. (2022). An efficient
MRI impulse noise multi-stage hybrid filter based on
cartesian genetic programming. In Advances in Natu-
ral Computation, Fuzzy Systems and Knowledge Dis-
covery, pages 95–106, Cham. Springer International
Publishing.
Kalkreuth, R. (2022). Phenotypic duplication and inversion
in cartesian genetic programming applied to boolean
function learning. In Proceedings of the Genetic and
Evolutionary Computation Conference Companion,
GECCO ’22, page 566–569, New York, NY, USA. As-
sociation for Computing Machinery.
Kaufmann, P. and Kalkreuth, R. (2017). An empirical study
on the parametrization of cartesian genetic program-
ming. In Proceedings of the Genetic and Evolutionary
Computation Conference Companion, GECCO ’17,
page 231–232, New York, NY, USA. Association for
Computing Machinery.
Kaufmann, P. and Kalkreuth, R. (2020). On the parame-
terization of cartesian genetic programming. In 2020
IEEE Congress on Evolutionary Computation (CEC),
pages 1–8.
Kruschke, J. K. (2013). Bayesian estimation supersedes the
t test. Journal of Experimental Psychology: General,
142(2):573.
Leitner, J., Harding, S., F
¨
orster, A., and Schmidhuber,
J. (2012). Mars terrain image classification using
cartesian genetic programming. In Proceedings of
the 11th International Symposium on Artificial Intel-
ligence, Robotics and Automation in Space, i-SAIRAS
2012, pages 1–8. European Space Agency (ESA).
Manazir, A. and Raza, K. (2022). pcgp: A parallel imple-
mentation of cartesian genetic programming for com-
binatorial circuit design and time-series prediction. In
2022 International Conference on Electrical, Com-
puter and Energy Technologies (ICECET), pages 1–4.
Margraf, A., Stein, A., Engstler, L., Geinitz, S., and Hah-
ner, J. (2017). An evolutionary learning approach to
self-configuring image pipelines in the context of car-
bon fiber fault detection. In 2017 16th IEEE Interna-
tional Conference on Machine Learning and Applica-
tions (ICMLA), pages 147–154. IEEE.
Equidistant Reorder Operator for Cartesian Genetic Programming
73

Miller, J. F. (1999). An empirical study of the efficiency of
learning boolean functions using a cartesian genetic
programming approach. In Proceedings of the 1st An-
nual Conference on Genetic and Evolutionary Com-
putation - Volume 2, GECCO’99, page 1135–1142,
San Francisco, CA, USA. Morgan Kaufmann Publish-
ers Inc.
Miller, J. F. (2011). Cartesian Genetic Programming.
Springer Berlin Heidelberg, Berlin, Heidelberg.
Miller, J. F. (2020). Cartesian genetic programming: its sta-
tus and future. Genetic Programming and Evolvable
Machines, 21(1):129–168.
Payne, A. J. and Stepney, S. (2009). Representation and
structural biases in cgp. In 2009 IEEE Congress on
Evolutionary Computation, pages 1064–1071.
P
¨
atzel, D. (2023). cmpbayes. Available at:
https://github.com/dpaetzel/cmpbayes, commit =
4de0abc37ee28b35267db173d32bb96ca9e69236.
Suganuma, M., Kobayashi, M., Shirakawa, S., and Nagao,
T. (2020). Evolution of Deep Convolutional Neu-
ral Networks Using Cartesian Genetic Programming.
Evolutionary Computation, 28(1):141–163.
Torabi, A., Sharifi, A., and Teshnehlab, M. (2022). Us-
ing cartesian genetic programming approach with new
crossover technique to design convolutional neural
networks. Neural Processing Letters.
Turner, A. J. and Miller, J. F. (2015). Neutral genetic
drift: an investigation using cartesian genetic pro-
gramming. Genetic Programming and Evolvable Ma-
chines, 16(4):531–558.
Vassilev, V. K. and Miller, J. F. (2000). The advantages
of landscape neutrality in digital circuit evolution. In
Evolvable Systems: From Biology to Hardware, pages
252–263, Berlin, Heidelberg. Springer Berlin Heidel-
berg.
Walker, J. A. and Miller, J. F. (2004). Evolution and acquisi-
tion of modules in cartesian genetic programming. In
Genetic Programming, pages 187–197, Berlin, Hei-
delberg. Springer Berlin Heidelberg.
Walker, J. A. and Miller, J. F. (2008). The automatic ac-
quisition, evolution and reuse of modules in cartesian
genetic programming. IEEE Transactions on Evolu-
tionary Computation, 12(4):397–417.
White, D., Mcdermott, J., Castelli, M., Manzoni, L., Gold-
man, B., Kronberger, G., Ja
´
skowski, W., O’Reilly, U.-
M., and Luke, S. (2013). Better gp benchmarks: Com-
munity survey results and proposals. Genetic Pro-
gramming and Evolvable Machines, 14:3–29.
Wilson, D. G., Miller, J. F., Cussat-Blanc, S., and Luga,
H. (2018). Positional cartesian genetic programming.
arXiv preprint arXiv:1810.04119.
Yu, T. and Miller, J. (2001). Neutrality and the evolvability
of boolean function landscape. In Genetic Program-
ming, pages 204–217, Berlin, Heidelberg. Springer
Berlin Heidelberg.
ECTA 2023 - 15th International Conference on Evolutionary Computation Theory and Applications
74
