
A NEW GENERATION OF DIGITAL LIBRARY TO SUPPORT
DRUG DISCOVERY RESEARCH
Edy S. Liongosari, Anatole V. Gershman, Mitu Singh
Accenture Technology Labs, 161 N. Clark Street, Chicago, Illinois 60601, USA
Keywords: Data Integration, Visualization, Link Analysis, Drug Discovery.
Abstract: The recent explosion of publicly available biomedical information gave drug discovery researchers
unprecedented access to a wide variety of online repositories, but the sheer volume of the available data
diminishes its utility. This is compounded by the fact that these repositories suffer from a silo effect: data
from one cannot be easily linked to data in another. This is true for both publicly available sources and
internal sources such as project reports. The ability to explore all aspects of biological data and to link data
across sources is beneficial, as it allows researchers to discover new knowledge and to identify new
collaboration opportunities by exploiting links. This paper presents an approach to solving this problem and
an application that allows researchers to browse and analyze disparate bio-medical repositories as one
semantically integrated knowledge space.
1 INTRODUCTION
In the last two years, we have interviewed eighteen
drug discovery scientists from several
pharmaceutical companies and research institutes in
Europe and North America to better understand their
research tasks and information needs. These
scientists are responsible for identifying new
chemical compounds that have therapeutic purposes.
They spend between 20 and 90 percent of their time
reading scientific articles that might be pertinent to
their projects. In some cases, they scan over 900
abstracts and read 200 articles in a month just to
keep up. This translates to 45 abstracts and 10 full
articles a day – a very time consuming activity.
Until very recently, their primary external source
of information was MEDLINE (Katcher, 1999),
which contains over twelve million bibliographic
citations and abstracts from articles published in
over 4,600 bio-medical journals. However, with the
recent advancement of computational molecular
biology fields such as genomics and proteomics,
these scientists find an increasing number of new,
more structured information sources indispensable
(Elmasri & Navathe, 1999; National Library of
Medicine, 2003). Some of these sources include
GenBank (gene sequence), KEGG (biological
pathways) and OMIM (genetic disorders).
Furthermore, as pharmaceutical companies introduce
their own corporate intranets, highly valuable
internal information such as project reports, lab
notes and screening results become available
enterprise-wide and may be relevant to these
scientists.
As a result, these scientists have to face dozens
of information sources each with its own intricacies,
access methods and nomenclature. Even a simple
question such as “Is there any internal expert on T4
Polynucleotide Kinase?” can pose significant
challenges as no single source may contain the
answer. The answer may have to be constructed by
examining the authors of various articles, reports
and patent documents that are indirectly related to
this particular kinase, such as through its proteins
and biological pathways. While doing this is
certainly possible, it could be very time-consuming
as it requires access to multiple heterogeneous
internal and external sources.
Existing systems such as GeneLynx (Lambrix &
Jakoniene, 2003), DiscoveryLink (Haas et al., 2001)
and SRS (Etzold, Ulyanov & Argos, 1996) view this
as a distributed database problem and approach it by
integrating the underlying schemas of the sources
into a global schema and by providing a query
language to go against the schema. Few of them deal
with the nomenclature issues and the fact that
scientists may pose questions at a different level of
abstraction than what is available in the underlying
sources (Geffner et al., 1999).
301
S. Liongosari E., V. Gershman A. and Singh M. (2004).
A NEW GENERATION OF DIGITAL LIBRARY TO SUPPORT DRUG DISCOVERY RESEARCH.
In Proceedings of the Sixth International Conference on Enterprise Information Systems, pages 301-306
Copyright
c
SciTePress
Developing a system that is appealing to the
scientists is further complicated by their lack of
computer skills making direct exposure to a formal
query language, for example, impractical. Form-
based user interfaces are not effective as different
users with diverse backgrounds fill out the forms
differently.
In this paper, we introduce our approach to
addressing these issues and the Knowledge
Discovery Tool, an application that embodies our
answers. Our approach is based on the creation of a
semantic index to information contained in the
underlying heterogeneous sources. The basis for this
index is the knowledge model that relates all major
concepts in the domain. Domain and application-
specific rules utilize this index to infer relationships
of potential value to researchers. The tool’s
visualization framework enables intelligent
browsing to support research and investigation tasks
by providing the ability to uncover indirect or
hidden linkages among pieces of information.
2 KNOWLEDGE MODELING
Central to our approach is a technique for modeling
the kind of knowledge a scientist needs to perform
his or her job (Brody et al., 1999). This model
contains a representation of bio-medical concepts:
entities and the relationships between them. This
model is an ontology designed specifically for
scientists performing a predefined set of tasks.
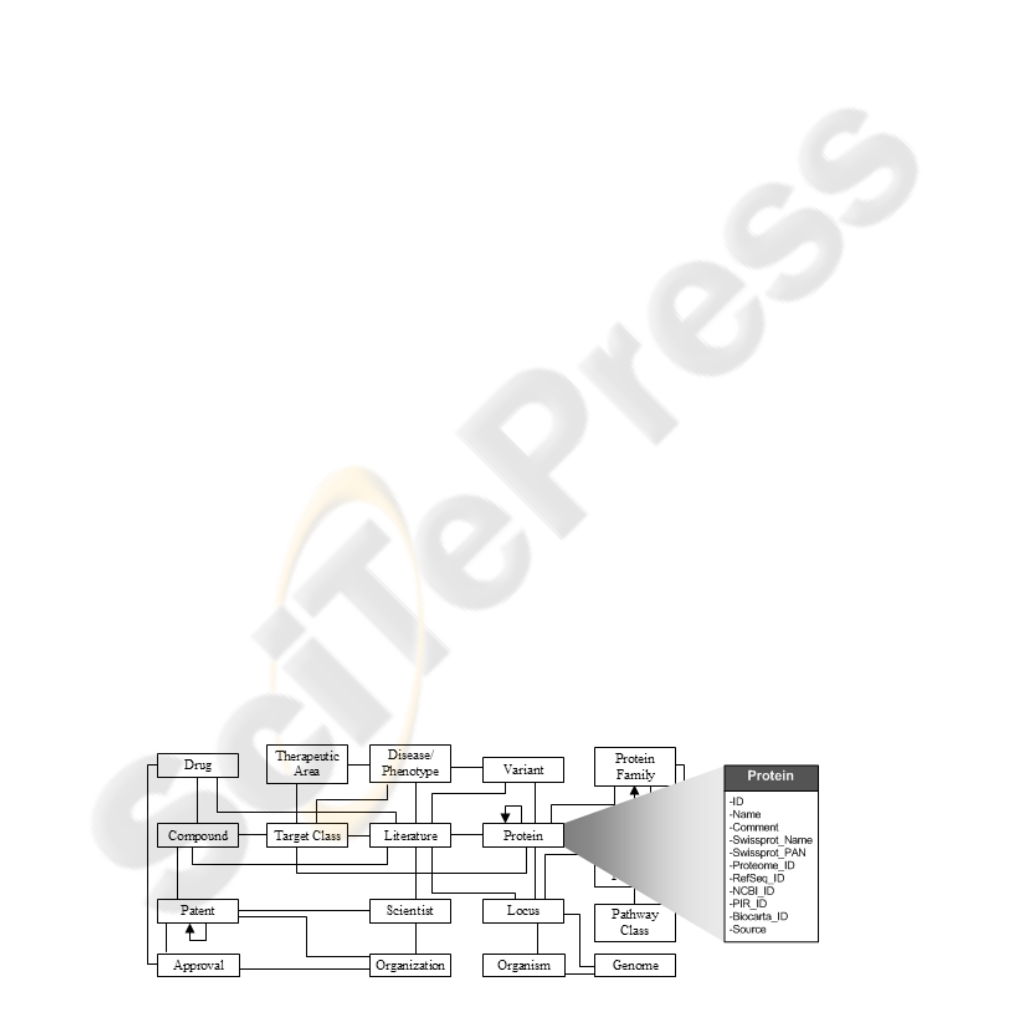
The model shown in Figure 1, contains entities
and relationships that depict treatments for a disease,
how those treatments are related to a set of drugs,
the chemical compounds that comprise those drugs,
how the compounds are related to various target
proteins, and so on. This knowledge model is a
representation of how the scientists think about the
information they need in order to perform their
tasks.
The model enhances the accessibility of
knowledge in three major ways. First, it creates a
layer that is independent of the location of the
underlying information. Second, the instantiated
model allows the user to search and browse the
entire body of knowledge as one homogeneous
space of related entities while maintaining links back
to the original sources.
Third, when used with classification hierarchies,
the model becomes a powerful abstraction
mechanism (Geffner et al., 1999). For example,
when a scientist inquires about the role of G-protein
in Central Nervous System (CNS) diseases, a
straight text search might not reveal anything as the
underlying sources may not contain the phrase
“Central Nervous System” or its synonyms.
However, these sources may contain references to
the relationship of G-protein and Parkinson’s or
Alzheimer’s diseases. To answer the above question,
the system must utilize a disease classification
hierarchy such as the one in Medical Subject
Headings (Lowe & Barnett, 1994) which classifies
Parkinson’s and Alzheimer’s as sub-diseases of
CNS. As a result, the system can exploit these
linkages and provide information about the
relationship between G-protein and higher level
diseases.
In addition to structural relationships, our
knowledge model contains dozens of domain-
specific functional rules such as “Any person who
has authored more than X documents on a protein is
an expert in that protein.” Such rules constrain and
guide the automatic inference process.
3 KNOWLEDGE DISCOVERY
TOOL
When the model is instantiated with existing data
sources as described in section 4, it becomes a giant
semantic index into the underlying sources and can
support a variety of applications. One can easily
build an SQL interface for example, to query the
index. However, given that our target users have
Figure 1: A knowledge model to support drug discovery scientists.
ICEIS 2004 - HUMAN-COMPUTER INTERACTION
302

little or no SQL or information retrieval skills, we
built a web-based intelligent browser called
Knowledge Discovery Tool (KDT) that uses our
semantic index and the model’s inference rules to
select and present the most likely relationships
among the data of interest to the user.
To demonstrate some of the KDT features,
consider the following interaction: A drug discovery
scientist who specializes in Oncology is researching
a potential link between Parkinson’s disease and
cancer. She needs to bring herself up-to-speed about
Parkinson’s disease in a short time. She needs to
identify experts in the area and any past work that
might be relevant.
She starts up KDT, selects the search type
“Disease” and types “Parkinson’s” into the search
box. KDT then displays all diseases whose names
include “Parkinson’s”. She clicks on “Parkinson’s
disease” and is brought to the Landscape View for
that disease (Figure 2). The view divides the screen
into 9 (3x3) panes. The center pane shows her
current focus (Parkinson’s Disease). Each outer pane
displays information related to the current focus as
filtered by the knowledge model. She sees recent
literature related to the disease in the top left pane, a
list of experts on the disease in the top center pane,
and a list of organizations that have published
articles or own patents related to the disease in the
top right pane. The center row contains a list of
related chemical compounds; the disease itself; and a
list of biological target classes used for treatments of
the disease.
The researcher can see linkages among the items
in the panes indicated by light grey lines that are
visible without cluttering the screen. The lines let
users visually maintain the connectivity among
entities, which seems to be intuitive to many users.
Studies have shown that exposing a large number of
relationships stimulates fresh thoughts and breaks
through creative blocks (Schneiderman, 2000).
The view facilitates fast browsing by minimizing
the need for mouse clicking. For example, if the user
hovers the mouse over an item, a tool-tip will pop up
displaying the item’s full title and attributes and it
will also highlight its links. Since each item
represents an index to the underlying source, the
user can view the source by double clicking on the
item.
Single clicking on an item will re-orient the
knowledge model, move the item to the center pane,
and refresh the contents of the outer panes
accordingly. Figure 3a shows how the view looks
(a) (b)
Figure 3: (a) Landscape view of gene “PRKN”, (b) A window showing a path that substantiates the link
between PRKN and UBE2L1
Figure 2: Landscape View of “Parkinson’s disease”.
A NEW GENERATION OF DIGITAL LIBRARY TO SUPPORT DRUG DISCOVERY RESEARCH
303

after the user has clicked on PRKN - the third gene
(locus) in the bottom middle pane of Figure 2.
Each outer pane of the browser presents the
results of a query run against the underlying
semantic index, filtered and prioritized according to
the rules in the knowledge model. The user can
easily customize the view in each pane by picking
from a large library of predefined views. For
example, the user can change the list of disease
expert shown in top center pane of Figure 3a to
show those people that have been published about
PRKN in the past 3 years and sort the list based on
their most recent publication date. A user who is an
oncologist can tailor a pane to display only genes
that relate to PRKN and cancer-related diseases.
KDT provides utilities to explore potential
linkages among entities that are depicted as grey
lines. For example, one of the items in the left center
pane in figure 3a, UBE2L1, is a gene that has been
associated with several forms of cancer including
Leukemia and Breast Cancer (Ardley et al., 2000).
The link between PRKN and UBE2L1 is drawn as a
dashed line indicating there are multiple degrees of
separation between them. By clicking on the right
menu over the line, the user can query the tool to
show how this link is derived. The tool found two
paths that substantiate the link. Figure 3b shows one
of them: a path with six items as follows. Gene
PRKN has been renamed to PARK2 and this gene
produces the Parkin protein. Parkin is linked to
another protein UbcR7 through two articles. The
bottom half of figure 3b shows one of the articles
that describes an interaction between Parkin and
UbcR7 in rat brain (Wang et al., 2001). UbcR7 in
turn is produced by UBE2L1. Deriving links in this
manner to expose hidden or indirect links that the
users might not have thought of is a powerful
capability (Schneiderman, 2000).
KDT also serves as a powerful collaboration
tool. By annotating items or links, users can
informally share their opinions and enrich the
existing contents. Furthermore, by continuously
monitoring common bookmarks and users’
navigation paths, the tool also can match users with
similar interests.
Because the index is continuously updated, the
researchers can use KDT to monitor new items and
links related to a topic of interest. We also
developed wizards similar to those used in setting up
printers in Microsoft Windows, to automate the
repetitive tasks that the users frequently do with
KDT.
4 INSTANTIATING THE MODEL
The process used to instantiate the model is very
similar to the one used for data warehousing
(Fayyad et al., 1996). The data from the selected
data sources is extracted and transformed into its
relational form. It is then cleansed of errors using a
semi-automated approach. Thesauri are created in
the process. Cleansed data is then integrated. This
process is described more in detail below.
4.1 Data Selection, Mapping and
Extraction
The 13 sources used in the current implementation
of KDT are: Enzyme, GeneOntology, NCBI
Genome, Interpro, KEGG, LocusLink, MeSH, NLM
Taxonomy, OMIM, MEDLINE, NCBI RefSeq,
Swissprot, and Unigene.
First, data fields in each source are mapped onto
the attributes of entities in our model. For example,
NLMTaxonomy’s TaxID, LocusLink’s OrganismID
and RefSeq’s OrganismID are all mapped to the
OrganismID in the model. We map all attributes of
all entities and relationships defined in our model.
As the above example indicates, most attributes in
the knowledge model are instantiated from multiple
sources. This, of course, generates conflicts and
they will be addressed in section 4.3.
Second, we create a representation of each
source in a local database. Once the information is in
the database, some of the fields have to be parsed
further. For example, the pathway description field
from KEGG may contain a piece of text like
“Glycolysis/Gluconeogenesis – Aquifex aeolicus”
which would be parsed into two subfields:
“Glycolysis/Gluconeogenesis” as the pathway’s title
and “Aquifex aeolicus” as the associated organism
name. This secondary parsing could also result in
building new hierarchies. For example, by parsing
the ExPASy’s EnzymeID, one can determine that
the protein family with EnzymeID of “EC.2.7.1.12”
is a child of EnzymeID “EC.2.7.1.” We use dozens
of rules to guide data extraction.
4.2 Schema Integration
This phase reconciles diverse schemas in the local
database with the schema as defined in the
knowledge model. Lenzerini (2002) describes this as
the local-as-view (LAV) process. It is done in two
steps. The first step handles the fact that a data
source could be mapped into multiple entities in the
knowledge model. This is accomplished by creating
multiple database views. The second step handles
ICEIS 2004 - HUMAN-COMPUTER INTERACTION
304

the fact that an attribute of an entity can be
assembled from multiple sources. Thus we need to
combine multiple database views from step one.
This is accomplished by writing SQL scripts to
insert the contents from the appropriate views to the
target table for each entity.
4.3 Instance Integration
Schema integration produces a large number of
redundant instances for each entity primarily due to
the fact that different sources use different
nomenclature or provide different set of attributes.
The objective of instance integration is to remove
conflicts and merge redundant data of an instance by
comparing one or more of its attributes.
We start instance integration by identifying
redundant records. We employ the vector space
model (Fasulo, 1999) to determine the similarity
among attributes through the use of two broad
classes of heuristics: ID-based and text-based. In ID-
based heuristics, we assume that there are one or
more IDs that uniquely identify an instance. While
this is the most straightforward in most cases, the
process is complicated by the fact that some of these
IDs may not be consistent.
The heuristics to solve ID consistencies were
manually developed by domain experts after visually
inspecting the origins of the inconsistencies. For
example, we found that the combination of
SwissprotID and OrganismID can be used to
uniquely identify a gene for our purpose. This class
of heuristics is applicable to gene, proteins,
pathways, protein families and genomes where the
use of IDs is quite pervasive.
The text-based heuristics are applicable to
organisms, phenotypes, organizations and people
where IDs are not readily available. We employ
many techniques and heuristics to resolve the name
similarity problem. Many of such techniques are
also used in WHIRL (Cohen, 2000). They range
from simple removal of punctuation (e.g.,
“Legionnaires' Disease” vs. “Legionnaires
Disease”), to comparing last name and first initial
(e.g., “A. aeolicus” vs. “Aquifex aeolicus”), to using
dictionaries and synonyms to match “zebrafish” to
“zebra danio”.
Our synonym tables are created in three ways.
First, there are several sources such as
NLMTaxonomy that contain synonym information
explicitly and we simply import them. Second, some
sources contain implicit synonym information that
we have to extract. For example, Swissprot’s protein
name may contain text like “Alzheimer's disease
amyloid A4 protein precursor (Fragment) (Protease
nexin-II) (PN-II) (APPI)“. From this piece of text we
extract “PN-II” and “APPI” as the synonyms of
“Protease nexin-II”. The third way is described in
conflict resolution below.
In conflict resolution, we tag each attribute with the
source from which the value was extracted. Each
source is associated with a confidence value between
0 and 10 by a domain expert. An attribute with a
higher confidence value can overwrite those with
lower confidence values. However, for the attributes
that signify the names of an object, we mark them as
potentially synonymous instead of overwriting them.
The merge step involves a domain expert to
confirm that the identified redundant records are
indeed redundant and that the suggested merged
record is correct. We developed a small application
called Thesaurus Builder to assist a domain expert in
this task. This application also allows the domain
expert to manually identify instances to merge.
While this manual step is time consuming, it is
important to have an expert to validate this step as
the quality of the entire integration result is highly
dependant on it. The decisions made by the domain
expert are captured so that they can be automatically
re-applied in the future.
4.4 Post-Integration
While published articles in MEDLINE form one of
the richest sources of bio-medical information to
date, automatically extracting semantic information
from them is hard (Jacquemin, 2001). Currently, we
use these articles to create weak unlabelled links
between the entities in the knowledge model through
the co-occurrences of terms in the articles. The more
articles that link the two entities, the stronger the
link is. The strength value is then used to rank and
sort the query results.
Another factor in determining the strength value
is the length of inferred relationships. For example, a
gene can be related to a disease through its proteins
and variants. As each intermediate step introduces
further uncertainty, we assigned lower strength to
the relationships inferred using longer paths.
5 OUTCOME AND BENEFIT
As shown in section 3, a KDT user is able to explore
thematically related information across multiple data
sources as if it were a single richly connected
knowledge space. The user does not have to be
concerned with the details of the underlying
databases, their formats, or their access methods.
Exploration of a potential link between two entities
as shown in Figure 3b that takes minutes with KDT
would require hours or perhaps days using the
A NEW GENERATION OF DIGITAL LIBRARY TO SUPPORT DRUG DISCOVERY RESEARCH
305

access methods provided by the 13 knowledge
sources we cover.
We developed the components used to
instantiate the knowledge model using Microsoft-
based technologies including Microsoft Windows
2000 Server and Microsoft SQL Database. KDT was
written entirely in Java as a Java applet. This eases
the deployment of the tool as it can run in most
Internet browsers that support Java.
From the thirteen sources we have selected, the
instantiated knowledge model contains over 1.1
million genes, 1.6 million proteins, 200,000
organisms, 12,000 pathways, 6,000 diseases, 12
million articles and over 4 billion relationships
across these biological entities. Prior to the post-
integration phase, the instantiated knowledge model
is about 70GB in size. Once the indices and pre-
calculated relationships are created, the size of the
database grows to 400GB.
While we have not conducted a formal
evaluation of KDT’s effectiveness as compared to
the current access methods, bio-medical researchers
who have tried KDT have given us very positive
feedback and expressed a strong desire to use the
tool in their daily activities. In one particularly
gratifying case, the senior leader of a heart failure
research project tried it during a ten-minute
demonstration and discovered a valuable
relationship between heart failure and leukemia of
which he had not been aware. In another case, a
post-doctoral biologist discovered a new link
between Lou Gehrig’s disease and genital diseases
suggesting that a drug for one disease can be
modified for the treatment of the other.
Currently, we are working with several
pharmaceutical companies, the University of
Colorado Health Sciences Center in Denver and the
Integrative Neuroscience Initiative on Alcoholism –
an initiative sponsored by the National Institute of
Alcohol Abuse and Alcoholism – to formally
evaluate the effectiveness of KDT. We have over 30
people who have signed up for our initial pilot. We
estimate the formal evaluation will be done in
Spring 2004.
REFERENCES
Ardley, H. C., Moynihan, T.P., Markham A.F. &
Robinson P.A., 2000. ‘Promoter analysis of the human
ubiquitin-conjugating enzyme gene family UBE1LI-4,
including UBE2L3 which encodes UbcH7’, Biochim
Biophys Acta, vol. 1491, no. 1-3, pp. 57-64.
Brody, A.B., Dempski, K.L., Kaplan, J.E., Kurth, S.W.,
Liongosari, E.S. & Swaminathan, K. S., 1999.
‘Integrating Disparate Knowledge Sources’, Proc.
Second Int. Conf. on Practical Application of
Knowledge Management, pp. 77-82.
Cohen, W.W., 2000. ‘Data integration using similarity
joins and a word-based information representation
language’, ACM Trans. Info. Systems, vol. 18, no. 3,
pp. 288-321.
Elmasri, R. & Navathe, S., 1999. ‘Genome Data
Management’, in Fundamentals of Database Systems,
Pearson Addison Wesley, 3rd edition, pp. 898-905.
Etzold, T., Ulyanov A. & Argos, P., 1996. ‘SRS:
information retrieval system for molecular biology
data banks’, Methods in Enzymology, vol. 226, pp.
114-128.
Fasulo, D.,
1999. Analysis on recent work on clustering
algorithms, Technical Report #01-03-02, Dept. of
Computer Science and Eng., U of Washington, Seattle.
Fayyad, U., Piatetsky-Shapiro, G. & Smyth, P., 1996.
‘The KDD Process for Extracting Useful Knowledge
from Volumes of Data’, Comm. ACM, vol. 39, no. 11
pp. 27-34.
Geffner, S., Agrawal, D., El Abbadi, A., & Smith, T.,
1999. ‘Browsing large digital library collections using
classification hierarchies’, Proc. Eighth Int. Conf. on
Info. and Knowledge Management, pp. 195-201.
Haas, L., Schwarz, P., Kodali, P., Kotlar, E., Rice, J. &
Swope, W., 2001. ‘DiscoveryLink: A system for
integrated access to life sciences data sources’, IBM
Systems Journal, vol. 40, no. 2, pp. 489-511.
Jacquemin, C., 2001. Spotting and discovering terms
through NLP, MIT Press, Cambridge, MA.
Katcher, B.S., 1999. MEDLINE: A Guide to Effective
Searching, Ashbury Press, San Francisco, CA.
Lambrix, P. & Jakoniene, V., 2003. ‘Towards transparent
access to multiple biological databanks’, Proc. First
Asia-Pacific Bioinformatics Conf., vol. 19, pp. 53-60.
Lenzerini, M., 2002. ‘Data Integration: A Theoretical
Perspective’, Proc. 21
st
ACM Symp. on Principles of
Database Systems, pp. 233 – 246.
Lowe, H. & Barnett, G., 1994. ‘Understanding and using
the medical subject headings (MESH) vocabulary to
perform literature searchers’, JAMA, vol. 271, pp.
1103-1108.
National Library of Medicine, 2003 (updated 7 Feb 2003).
Growth of GenBank. Retrieved 28 Jan 2004 from
http://www.ncbi.nlm.nih.gov/Genbank/
genbankstats.html
Schneiderman, B., 2000. ‘Creating Creativity: User
Interfaces for Supporting Innovation’, ACM Trans. On
Computer-Human Inter., vol. 7, no. 1, pp. 114-138.
Wang M., Suzuki, T., Kitadata, T., Asakawa, S.,
Minoshima, S., Shimizu, N., Tanaka, K., Mizuno, Y.
& Hattori, N., 2001. ‘Developmental changes in the
expression of parkin and UbcR7, a parkin-interacting
and ubiquitin-conjugating enzyme, in rat brain’, J.
Neurochemistry, vol. 77, no. 6, pp. 1561-1568.
ICEIS 2004 - HUMAN-COMPUTER INTERACTION
306
