
HOW MUCH SEQUENCE IDENTITY GUARANTEE GOOD
MODELS IN HOMOLOGY MODELING
Proteins from Serine Protease Family as a Test Case?
Jamal Riayn and Anwar Rayan
QRC-Qasemi Research Center, Al-Qasemi Academic College, P.O.B. 124, Baka El-Garbiah 30100, Israel
Keywords: Homology/comparative modeling, 3D-structure prediction, multiple sequence alignment.
Abstract: Homology modelling is utilized to predict the 3-D structure of a given protein (target) based on its sequence
alignment to a protein whose structure (template) has been experimentally determined. The use of such
technique is already rewarding and increasingly widespread in
biological research and drug
development. The accuracy of the predictions as commonly accepted is dependent on the score of
target protein - template sequence identity. Given the sequence identity score of pairs of proteins,
certain questions are raised as to whether we can assess or quantitate the quality of the obtained
model. Also, whether we should choose, the protein with the highest sequence identity as a
template. The answer to these questions is critical since only with such determinations, we could
decide how to choose the template and to which usage the model is reliable.
We intend in the
paper to assess the accuracy of sequence identity-based homology modeling by analyzing a
database of 4560 pair-wise sequence and structural alignments. The decision making process
regarding to which parts of the known protein to perform structural alignment is not trivial and
clearer rules should be extracted.
1 INTRODUCTION
The 3D structure determination of a certain protein
greatly helps unravelling its function and binding
mechanisms. Such structural information can also
aids in designing experiments in mutagenesis and
even utilized for structure-guided drug development
or virtual screening
1
. Since experimental structures
are available only for a small number of sequenced
proteins, alternative strategies are required to predict
reliable models for protein structures when X-ray
diffraction or NMR are not yet available
2
. Among
the different strategies currently used for
constructing 3-dimensional structures of certain
proteins, we shall find the homology modeling
(termed also as comparative modeling) as the most
accurate method among the computational methods,
yielding reliable models. Another approach termed
“ab-initio” modeling, is not practical yet for the
construction of reliable models
3
. According to the
state of art, a three dimensional template is chosen
by virtue of having the highest level of sequence
identity with the target sequence, and similar
secondary and tertiary structure (belongs to the same
"fold"). Baker and Sali
3
have shown that a homology
model for a protein at medium size at least and with
sequence identity of less than 30% to the template
crystal structure is unreliable. The rule of =>30% of
sequence identity score does not specify how
identity should be distributed along a sequence. The
quality of the models obtained by comparative
modeling is mostly quantitated by the root mean
square deviation of the backbone atoms or the
positions of alpha carbons (termed Cα RMSD)
between model and experimental structure. A model
can be considered 'accurate’ or ‘reliable’ model
when its RMSD is within certain spread of
deviations. How big is this spread?
The comparative modeling procedure for
developing a three-dimensional model from a
protein sequence based on the structure of
homologous protein is built generally from few
steps: after identifying the homologous protein and
performing optimal sequence alignment (based on
score of identity or similarity), the structurally
conserved regions (SCRs) are identified and
coordinates for the core of the models are generated.
Following the core generation, one predicts the
455
Raiyn J. and Rayan A. (2009).
HOW MUCH SEQUENCE IDENTITY GUARANTEE GOOD MODELS IN HOMOLOGY MODELING - Proteins from Serine Protease Family as a Test
Case?.
In Proceedings of the International Conference on Bio-inspired Systems and Signal Processing, pages 455-458
DOI: 10.5220/0001379204550458
Copyright
c
SciTePress

conformations of the structurally variable regions
(termed loops) and adds the side chains. Some
approaches, align multiple known structures firstly,
then, identifying structurally conserved regions to
construct an average structure, for modeling these
regions of the inquiry protein.
In this communication, we analyze a database of
pairs of proteins, sequence and structurally aligned
and raised few questions:
i. Can we predict the accuracy of the modelled
structure based on sequence identity score?
ii. When the selection of the protein with highest
identity score is justified?
iii. Can we formulate a set of rules for homology
modeling?
1.1 Materials and Methods
More than 124 unique homologs of the serine
protease family of proteins that have sequence
identity below 99% were downloaded from the
Brookhaven Protein Databank (PDB). Then, IMSA -
Intelligent Multiple Sequence Alignment
4
(in-house
software based on the Intelligent Learning Engine
(ILE) optimization technology) was utilized to
optimally align the whole set of all sequences.
Sequence identity score was calculated for each pair
of sequences. All residues from the multiple
sequence alignment were found only on 96 proteins
(see table 1). Other proteins lack coordinates of one
residue at least in their 3D structures. The alpha
carbons for residues of selected proteins were
extracted from the PDB structures and structurally
superimposed.
The quality of the models obtained by homology
modeling is quantitated with the Cα RMSD between
model and experimental structure. We have defined
'highly accurate’ model as one having <=2 Å RMSD
from the experimentally determined structure, while
models having Cα RMSD above this threshold and
<=4 Å were termed “reliable” models which could
fit for designing mutagenesis experiments but not
drug design and binding affinity tests. BioLib was
used for performing structural alignment and for
computing the Cα RMSD (BioLib is an open-
environment developing toolkit developed by
BioLog Technologies Ltd.).
The multiple sequence alignment matrix
obtained from running our in-house software on the
selected database of serine proteases, was processed
as described below, in order to specify which parts
of the whole set of sequences to select for homology
modeling. We use a “voting” approach, in which
each amino acid contributes to the conservation at a
sequence position according to its frequency in that
particular position (see equation 1). These
frequencies are measured in all sequences of the
database.
%100∗=
k
n
C
ij
ij
(1)
C
ij
is thus the conservation factor for residue type i
at sequence position j.
n
ij
is the number of
sequences, which have amino acid i at position j of
the multiple alignment, and k is the total number of
sequences in the database.
Table 1: PDB codes of 96 serine proteases (the first four
letters are the code of the protein in the PDB while the last
letter is the chain ID).
1AMHA 1ANB0 1ANC0 1AND0 1BRBE
1CO7E 1DPO0 1F7ZA 1SLUB 1SLWB
3TGJE 1QL9A 1J16A 1TRMA 1EZSC
1F5RA 1FY8E 3TGKE 1AN1E 1MCTA
1S83A 1TAWA 1UTNA 1OPHB 1V2OT
1V2QT 1V2RT 1V2ST 1V2WT 1V2NT
1V2LT 1H4WA 1TRNA 1UTMA 1HJ8A
1MBQA 1BIT0 1A0JA 1DX5M 1JOUB
1RD3B 1THPB 1C5LH 1H8DH 2THFB
1H8IH 1B7XB 1BTHH 1TQ7B 1SHHB
1VR1H 1UCYK 1EUFA 1FI8A 1PJPA
1NN6A 1KLT0 1IAUA 1GVKB 1HAXB
1QNJA 1BRUP 1DST0 1BIO0 1RFNA
1PFXC 1A0LA 1CGHA 1FXYA 1LO6A
1G2LA 1FAXA 1LTOA 1TON0 1NPMA
1MZAA 3RP2A 1AO5A 1KLIH 1KIGH
1AZZA 1EAXA 1GVZA 1PYTD 1OP8A
1ORFA 1RTFB 1AUTC 1P57B 1FIZA
1FIWA 1BQYA 1A5IA 1MD8A 1EQ9A
1EKBB
2 RESULTS AND DISCUSSION
In this study, we aim to assess models obtained by
homology protein modeling by looking on a large
set of sequence/structure alignments that belong to
the same protein family (adopt the same “fold”). We
have used in-house software for multiple sequence
alignment and the regions for model construction
(firstly using all the Cα atoms of the 160 common
residues and at the second time, we chose for model
construction SCRs based on the structural analysis
of one protein (1A0JA), see figure 1. The pair-wise
sequence alignments in our database ranges between
28% and 100%.
Sequence analysis of the database revealed
highly conserved amino acids that where distributed
along the protein chain (see figure 1, number of
BIOSIGNALS 2009 - International Conference on Bio-inspired Systems and Signal Processing
456
amino acids found above certain conservation
thresholds, and see table 2, residues with
conservation threshold above 95% as an example).
We expect that those residues in there spatial
coordinates play important role in the protein
function and/or in stabilizing the protein folding (or
conformation). Thus, the inter-residue distance
matrix should be somehow similar in each protein.
This could be assessed qualitatively by extracting
those residues from the x-ray structures of the
proteins and performing pair-wise superposition. As
depicted in table 3, the Cα RMS deviation is very
low in average in all pairs. These results reveal the
correctness of the multiple sequence alignment and
could be used in model refinement of serine
proteases. The averaged root mean square coordinate
deviation correlates well with the percentage identity
within the highly conserved residues with
correlation coefficient of 0.9695. 4560 models of
proteins were generated and as depicted on figure 2,
when the sequence identity with the template is
>60%, the constructed model is always highly
accurate, while when the sequence identity is less
than 50% models based on templates with sequence
identity less than protein with the highest score
should be assessed. We have reached the same
conclusion when analysing parts of the proteins
including variable regions (loops). Since the
methods for predicting the loop conformations and
not yet highly accurate, we should model them based
on the template structure in certain circumstances.
For all 160 residues in our multiple sequence
alignment models, we have computed the sequence
identity percentage between target and template
sequences and the RMSD of the models from their
corresponding experimental template. Although the
stretches of the models contain large parts from the
variable regions, we have obtained mostly reliable
models.
Mostly, models of secondary structure segments
that where built based on templates which share any
degree of sequence identity (> 28%) with the target
are highly accurate (table 3) and seem to be useful
for drug design and docking experiments. However,
when the degree of sequence identity is lower
than 50%, the best template to thread on is not
always the one with the highest identity score.
Other templates should be evaluated in order to get
more accurate models. We obtain higher percentage
of accuracy when we chose the best structured
protein to be used as a template, perform the correct
alignment and choose the correct stretches to
remodel. One of the major contributors to the
models inaccuracy could be performing the
wrong threading. Position conservation threshold
may be used for further refinement of the model
applying molecular dynamics (MD), simulated
annealing (SA), iterative stochastic elimination
(ISE) or other optimization approaches
5
.
Conservation ratio (160 residue positions sequence
aligned in 98 serine proteases)
R
2
= 0.9829
0
20
40
60
80
100
50 60 70 80 90 100
Positional identity percentage threshold
Conserved residues
Figure 1: Analysis of positional conservations in the
sequences of 96 unique serine proteases. Each protein has
160 residues and the multiple sequence alignment was
performed without gaps.
Table 2: Positional Conservation Threshold (PCT)
calculated according to equation 1.
PCT* Average RMSD Median
Standard
Deviation
100 0.503 0.463 0.219
95 0.631 0.486 0.387
90 0.621 0.497 0.361
85 0.609 0.492 0.342
80 0.704 0.577 0.368
75 0.757 0.702 0.361
70 0.812 0.822 0.362
65 0.862 0.898 0.372
60 0.894 0.920 0.405
55 0.936 0.979 0.405
50 0.990 1.059 0.408
* Position Conservation Threshold – the residue should be
conserved above this threshold in the certain position.
3 CONCLUSIONS
We present in this paper, sequence and structural
analysis of 4560 pairs of proteins and raise few
questions regarding the homology modeling
procedure. In view of the data above, the most
important question was whether the sequence
HOW MUCH SEQUENCE IDENTITY GUARANTEE GOOD MODELS IN HOMOLOGY MODELING - Proteins from
Serine Protease Family as a Test Case?
457

Table 3: Probabilities of modeling approach accuracy for
target-template identity classes in serine protease family.
Secondary structure segments were used for Root Mean
Square Deviation (RMSD) measurements.
Percent
sequence
identity
α
Total
number
of
models
β
Percent
π
models
with
RMSD <
1 Å
Percent
models
with
RMSD < 2
Å
Percent
models
with
RMSD <
3 Å
25-29 15 40 100 100
30-39 883 28 98 100
40-49 2365 50 99.9 100
50-59 423 75 100 100
60-69 51 90 100 100
70-79 181 100 100 100
80-89 289 100 100 100
90-95 44 100 100 100
α: Sequence identity range between target and template.
β: Total number of models in any given sequence identity range.
The table summarises 4251 model – template pairs.
π: Percent of models, in a given sequence identity range, deviates
by 1 Å or less from the corresponding experimental control
structure. The following columns provide these percantages for
other RMS deviations.
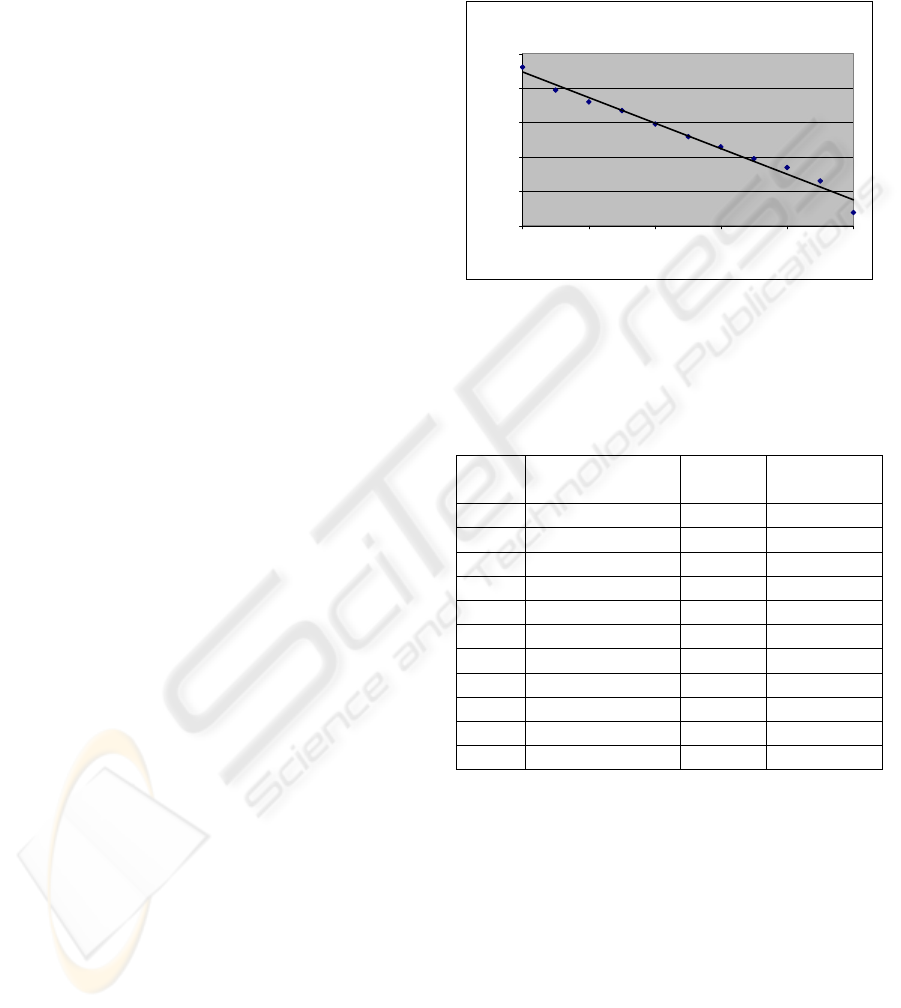
Figure 2: This plot describe the relationship between
RMSD and sequence identity percentage. We can
discriminate easily between surely good models when the
sequence identity is above 50-60% and models with high
uncertaintity when the sequence identity is less than 50%.
Each model contain all 160 residues.
identity score against all experimentally determined
structures of proteins will alone assist (or be
sufficient) in deciding which protein to use as the
template for the homology modeling and how to
improve the threading process. The results revealed
that when
the sequence identity with the template is
>60%, it is justified to select the protein with the
highest score as a template. While, when the
sequence identity is less than <50%, we should
select more than one template for assessing.
Alignment based on analysis of large database of
certain fold could give better results than those
obtained by optimized pair-wise alignment. Further
research and analysis of databases of proteins which
belong to other folds may aid us in formulating
clearer rules for the homology modeling process. As
well, usage of position conservation threshold in
model refinement is recommended and is currently
under evaluation in our lab.
REFERENCES
Patny A., Desai P.V., Avery M.A., 2006. Current
Medicinal Chemistry, 13(14), 1667-91
Eszter H., Zsolt B., 2008. Journal of Structural Biology,
162, 63-74
Baker D., Sali A., 2001. Science 294 (5540), 93-96
Rayan A.M., Raiyn J.A., 2008. Intelligent Learning
Engine (ILE) Optimization Technology, Provisional
Rayan A., Noy E., Chema D., Levitzki A., Goldblum A.,
2004. Current Medicinal Chemistry, 11, 675-692.
Sequence Identity versus RMS Deviation
(Secondary-structure based on 1aoj [code in
PDB])
0
1
2
3
20 40 60 80 100
Sequence Identity
RMSD
BIOSIGNALS 2009 - International Conference on Bio-inspired Systems and Signal Processing
458
