NOVEL APPROACHES FOR RETRIEVING PROTEIN 3D
STRUCTURES
1
Georgina Mirceva,
2
Ivana Cingovska,
3
Zoran Dimov
1,2
Faculty of Electrical Engineering and Information Technologies, Univ. Ss. Cyril and Methodius, Skopje, Macedonia
3
Microsoft Corporation, Vancouver BC, Canada
1
georgina@feit.ukim.edu.mk,
2
ivana.cingovska@feit.ukim.edu.mk,
3
zodimov@microsoft.com
4
Slobodan Kalajdziski,
5
Danco Davcev
4,5
Faculty of Electrical Engineering and Information Technologies, Univ. Ss. Cyril and Methodius, Skopje, Macedonia
4
skalaj@feit.ukim.edu.mk,
5
etfdav@feit.ukim.edu.mk
Keywords: Protein Data Bank (PDB), Protein retrieval, Discrete Fourier Transform, Discrete Wavelet Transform.
Abstract: To understand the structure-to-function relationship, life sciences researchers and biologists need to retrieve
similar structures from protein databases and classify them into the same protein fold. With the technology
innovation, the number of protein structures increases every day, so, retrieving structurally similar proteins
using current algorithms may take hours or even days. Therefore, improving the efficiency of protein
structure retrieval becomes an important research issue. In this paper, we propose three novel approaches for
retrieving protein 3D structures, which rely on the 3D structure of the proteins. In the first approach,
Discrete Fourier Transform is applied to protein structures. Additionally, some properties of the primary and
secondary structure of the protein are taken. In the second approach, some modification of the ray based
descriptor is applied on the backbone of the protein molecule. In the third approach, a wavelet transfor-
mation is applied on the distance matrix of the protein. The results show that the proposed ray based
descriptor gives the best average retrieval accuracy (92.95%), while it is much simpler and faster than the
other approaches.
1 INTRODUCTION
To understand the structure-to-function relationship,
life sciences researchers and biologists need to
retrieve similar structures from protein databases
and classify them into the same protein fold.
The
structure of a protein molecule is the main factor
which determines its chemical properties as well as
its function. Therefore, the 3D representation of a re-
sidue sequence and the way this sequence folds in
the 3D space are very important. The 3D protein
structures are stored in the world-wide repository
Protein Data Bank (PDB) (
Berman, 2000) which is
the primary repository for experimentally determi-
ned protein structures. With the technology innovati-
on the number of 3D protein structures increases
every day, so, retrieving structurally similar proteins
using current algorithms takes too long. Therefore,
improving the efficiency of protein structure
retrieval becomes an important research issue.
Many approaches for protein retrieval are based
on techniques for retrieving 3D objects. Since all the
algorithms in (Vranic, 2004) are applied for any kind
of 3D objects, but they are not yet applied for pro-
tein retrieval, we made some modifications of them
for building protein structure retrieval system.
Many protein retrieval systems use the distance
matrix as a representative of the protein’s 3D stru-
cture. The distance matrix is a symmetrical matrix
whose elements are the Euclidian distances between
Cα atoms. These matrices are treated as images and
different image processing techniques are performed
on them, leading to descriptors which are basis for
protein indexing. Even the well known DALI algo-
rithm uses distance matrix for structural alignment
of proteins (
Holm, 1996).
In (
Chi, 2004) the similarity between two images
that represent distance matrices is deduced by
directly analyzing their visual characteristics.
Namely, the image is divided in several bands

parallel to the main diagonal and the feature vector
of each image consists of a set of local visual
characteristics for each band as well as global visual
characteristics of the texture of the whole image,
like: uniformity of energy, entropy, homogeneity,
contrast, correlation, cluster tendency etc.
In (Marsolo, 2006) wavelet transformation on the
image is performed. The feature vector consists of
the approximation coefficients obtained at the fourth
level of Haar wavelet decomposition. At level four,
the feature vector has reasonable size of 36
coefficients, while the information about the global
structure is still preserved.
In this paper, we present three approaches for
retrieving protein. We have adopted the method
given in (Vranic, 2004) to extract the voxel protein
descriptor. Additionally, some properties of the pri-
mary and secondary structure of protein are taken as
in (
Daras., 2006), thus forming better integrated
descriptor. In the second approach, we incorporate
some modifications of the ray based descriptor and
apply it to the interpolated backbone of the protein
molecule. In the third approach, a wavelet
transformation is applied on the distance matrix of
the protein molecule. Finally, we present the
retrieval accuracy of our approaches. The evaluation
of the retrieval accuracy was made according to the
SCOP hierarchy (
Murzin, 1995). We provide some
experimental results of the analysis.
The proposed research approaches are given in
section 2, while in section 3 the experimental results
of the comparative analysis for retrieval accuracy are
presented. Section 4 concludes the paper and gives
some future work directions.
2 OUR RESEARCH
APPROACHES
The primary, secondary and tertiary structures of the
protein are stored in PDB. First, we will assume that
one protein is totally described by the arrangement
of its atoms in the 3D space. Each protein is made
up of several polypeptide chains. The protein back-
bone is built from Cα atoms. The alignment of these
atoms is relevant enough to describe the 3D structu-
re of the protein that will be shown later. We pro-
pose three approaches for retrieving protein molecu-
les which rely on the 3D structure of the protein.
2.1 Voxel Based Approach
We have used the voxel-based algorithm presented
in (Vranic, 2004) to extract the voxel descriptor. In
(Vranic, 2004), this algorithm is proposed for any
kind of objects, but it was not yet applied for protein
3D structure retrieval.
First, we extract the features of the tertiary stru-
cture of the protein by using the voxel based algori-
thm (Vranic, 2004). The surface of each atom is
triangulated, so forming 3D-mesh model of the
protein. After triangulation, we perform voxeliza-
tion. Voxelization transforms the continuous 3D-
space, into discrete 3D voxel space. Depending on
positions of the polygons of a 3D-mesh model, to
each voxel, a value is attributed equal to the fraction
of the total surface area of the mesh which is inside
the voxel. The information contained in a voxel grid
is processed further to obtain both correlated infor-
mation and more compact representation of voxel
attributes as a feature. We applied the 3D Discrete
Fourier Transform to obtain a spectral domain fea-
ture vector which provides rotation invariance. This
vector presents geometrical properties of the protein.
Additionally, some attributes of the primary and
secondary structure of the protein molecules are ex-
tracted as in (Daras, 2006). By incorporating these
features we provide better integrated descriptor.
The geometrical descriptors are compared in
pairs by using (1), as in (Vranic, 2004).
p
gg
R
gg
R
G
ffffD
''''''
min),(min
αα
αα
−==
∈∈
(1)
The similarity in primary and secondary structure
is evaluated by (2) where additionally different
weights to the features were assigned.
∑
=
−=
34
1
2
'''
)]()([
i
ssiS
ififWD
(2)
The overall similarity is determined by (3). As it
can be seen, our algorithm is mainly based on
geometrical features.
D= k
1
D
G
+ k
2
D
S ,
k
1
=90%, k
2
=10%
(3)
2.2 Ray Based Approach
Cα atoms form the backbone of the protein mole-
cule. Analyses showed that by taking into account
only the Cα atoms of the protein and extracting
some suitable descriptor, we can get better results.
Proteins have distinct number of Cα atoms. So,
we have to find a unique way to represent all
proteins with descriptors with same length. In this
approach, we approximate the backbone of the
protein with fixed number of points, which are
equidistant along the backbone.
Finally, we use modification of the ray based
descriptor (Vranic, 2004).
In the process of retrieval, descriptors are
compared by using the L
1
and L
2
norm.
2.3 Wavelet Based Approach
In this approach, we used similar scheme as given in
(Marsolo, 2006). First, the distance matrix is calcu-
lated. The distance matrix is very good represen-
tation of the protein 3D structure, namely, proteins
with similar structure will have similar distance
matrices. Also, distance matrix is invariant of
scaling, translation and rotation of the protein.
However, the distance matrix can not be used as
a descriptor, because calculation of the distance
between two descriptors of that size will have
complexity O(n
2
). Thus, in this approach a wavelet
analysis of the distance matrix is performed.
The wavelet transform is very useful and popular
tool in signal processing. Its main advantage is that
it can provide analysis of an image in different
resolution, and unlike the Fourier transform, not
only the frequency components of the image are
obtained, but they are also localized in space.
Since the discrete wavelet transform can be
applied only to signals of length 2
n
, the distance
matrix is scaled to nearest upper 2
n
by using techni-
ques for image scaling. This can also be done by
interpolating the 3D skeleton with additional Cα
atoms up to some predefined number of type 2
n
. The
obtained distance matrix will have size 2
n
x2
n
.
The detail coefficients, which represent the high
frequency components of the signal, are obtained by
filtering the signal with high-pass filter. Similarly,
the approximation coefficients, which represent the
low-frequency components of the signal, are
obtained by filtering the signal with low-pass filter.
The filtering of the signals is performed by convol-
ving the signal with the impulse response of the
high-pass filter and the low-pass filter of the particu-
lar wavelet transform for the details and approxima-
tion coefficients respectively.
In this paper the wavelet analysis goes to the last,
n-th level of decomposition (if the size of the matrix
is 2
n
), i.e. to the minimal resolution of the image
which is one approximation coefficient (pixel). That
coefficient will represent the average value of the in-
tensity of the image. If the feature vector consists of
all the obtained wavelet coefficients, then
performance of the comparison will not be changed.
So, the feature vector must be some subset of the
wavelet coefficients. The high-frequency
coefficients of the wavelet transform carriers the
signal details. Thus, those coefficients are not
relevant for the descriptor, because they will
represent the local protein 3D structure
characteristics. The structural protein comparison
means comparing their global structural characte-
ristics. That means that the low-frequency coeffici-
ents should represent the protein. In our experiment,
we have used 20 - 255 biggest wavelet coefficients.
Additionally, they are quantized to values 1 (positive
ones) and -1 (negative ones). In this paper we use
Haar wavelet transform.
By observing the distance matrices seen as
images, it can be noticed that they are divided in
regions with same or similar colour, which means
that the low-frequency components dominate in the
image. Since the Haar wavelet is good representative
of regions with low-frequency, it is expected that
this wavelet will retrieve similar images with high
precision. The length of the Haar filter banks is 2, so
the calculation of the Haar wavelet transform will be
very efficient.
In the process of retrieval, let with Q[i,j] and
T[i,j] label the coefficient in the wavelet matrices on
position [i,j]. Suppose that Q[0,0]= T[0,0]=0. For
matrices with dimensions mxn, we can use (4) to
measure their distance. The weights w
i,j
are
empirically obtained, and because the main
information of the image is in the upper-left corner,
the limitation w
i,j
=w
min
(max(i,j),5) is performed. To
speed up the time complexity of the function d(Q,T),
we only look for the coefficients that are on the
same location.
∑∑
==
−
−−
=
n
i
m
j
ji
w
TQwTQd
00
)5),,min(max(
0,0
|]0,0[]0,0[|),(
(4)
3 EXPERIMENTAL RESULTS
We have implemented a system for protein retrieval
based on the three approaches described above. Our
ground truth data contains 6979 randomly selected
protein chains from 150 SCOP protein domains.
90% of the data set serves as the training data and
the other 10% serves as the testing data. We will
examine the retrieval accuracy of the descriptors
according to SCOP hierarchy (
Murzin, 1995).
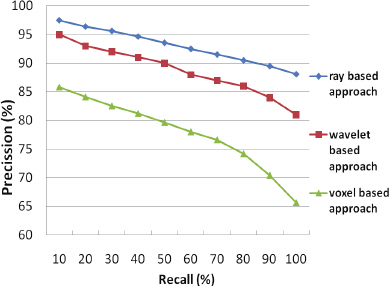
Figure 1: Retrieval results of our approaches.
Figure 1 shows the precision-recall diagrams of
our approaches. As can be seen from Figure 1, the
ray-based descriptor gives the best retrieval accuracy
(92.95%), while it is much simpler and faster than
the other descriptors.
4 CONCLUSIONS
In this paper we propose three novel approaches for
protein molecules retrieval. All approaches rely on
3D structure of protein molecules.
A part of the SCOP 1.73 database, which pro-
vides a structural classification of the proteins, was
used to evaluate the retrieval accuracy. The results
show that the ray-based descriptor gives the best
retrieval accuracy. The ray-based descriptor is also
much simpler and faster than other approaches. We
provide some experimental results.
The results showed that it is much better to take
into account only Cα atoms of the protein for extra-
cting protein descriptor (as in ray and wavelet based
approach which are more accurate than the other
approaches which take into account the whole 3D
structure).
Our future work will be concentrated on investi-
gating more efficient descriptors by incorporating
some additional features to descriptors.
REFERENCES
Berman, H. M., Westbrook, J., Feng , Z., Gilliland, G.,
Bhat, T. N., Weissig, H., Shindyalov, I., N., Bourne,
P., E., 2000. The Protein Data Bank. Nucleic Acids
Research, 28(1), 235-242.
Chi, P. H., Scott, G., Shyu, C. R., 2004. A Fast Protein
Structure Retrieval System Using Image-Based
Distance Matrices and Multidimensional Index, In
BIBE'04, Fourth IEEE Symposium on Bioinformatics
and Bioengineering, pp. 522-532.
Daras, P., Zarpalas, D., Axenopoulos, A., Tzovaras, D.,
Strintzis, M.,G., 2006. Three-Dimensional Shape-
Structure Comparison Method for Protein Classi-
fication. IEEE/ACM Transactions on computational
biology and bioinformatics, 3(3). pp. 193-207.
Holm, L., Sander, C., 1996. The FSSP Database: Fold Cla-
ssification Based on Structure-Structure Alignment of
Proteins. Nucleic Acids Research, 24(1), pp. 206-209.
Marsolo, K., Srinivasan, P., Ramamohanarao, K., 2006.
Structure-Based Querying of Proteins Using Wavelets.
In CIKM’06, ACM Fifteenth Conference on
Information and Knowledge Management, pp. 24-33.
Arlington, USA.
Murzin, A. G., Brenner, S. E., Hubbard, T., Chothia, C.,
1995. Scop: A Structural Classification of Proteins
Database for the Investigation of Sequences and
Structures. Journal of Molecular Biology, 247, pp.
536-540.
Vranic, D. V., 2004. 3D Model Retrieval. Ph.D. Thesis.
University of Leipzig.
