
IMPROVED DISEASE OUTCOME PREDICTION BASED ON
MICROARRAY AND CLINICAL DATA COMBINATION AND
PRE-VALIDATION
Jana
ˇ
Silhav´a and Pavel Smrˇz
Faculty of Information Technology, Brno University of Technology, Boˇzetˇechova 2, 61266 Brno, Czech Republic
Keywords:
Boosting, Clinical data, Combined models, Generalized linear models, Logistic regression, Microarray data,
Model evaluation.
Abstract:
Combining relevant information from high-dimensional microarray data and low-dimensional clinical vari-
ables to predict disease outcome is important to improve treatment decisions. Such a combination may yield
more accurate predictions than those obtained based on the use of microarray or clinical data alone. We pro-
pose a combination of logistic regression for clinical data and BinomialBoosting for microarray data. Then
we propose its extension designed for redundant sets of data. Our approach combines microarray and clinical
data at the level of decision integration. The extension includes pre-validation of models built with microarray
and clinical data followed by weights calculation. Weights determine relevance of microarray and clinical
models for data combination. Evaluations are performed with several redundant and non-redundant simu-
lated datasets. Then some tests are applied to two real benchmark datasets. Our approach increases outcome
prediction on non-redundant simulated datasets and does not decrease outcome prediction on redundant sim-
ulated datasets. Pre-validation of built models improves outcome of the prediction up to 4% in the case of real
redundant dataset.
1 INTRODUCTION
Clinical variables such as tumor grade, tumor size,
age, gender, family history and others depending on
the type of cancer have been used in prediction of
disease status and progression (Gajdos et al., 1999).
On the other hand, microarray data is an alternative
way of disease prediction (Michiels et al., 2005; Klijn
et al., 2005). Combining relevant information from
high-dimensional genomic data and low-dimensional
clinical variables to predict disease outcome is im-
portant to improve treatment decisions. Data combi-
nation increases prediction accuracy and may derive
a hybrid prognostic signature from combined data.
Attractiveness of prediction problems that can in-
clude disease outcome prediction or survival analysis,
comes from their ability to identify a group of patients
that can avoid aggressive chemotherapy (Fernandez-
Teijeiro et al., 2004).
Here, disease outcome is defined as a variable that
can have two values: poor prognosis or good progno-
sis, so we focus on a binary class prediction. The class
prediction is classification where the algorithm learns
from samples with known class membership (training
set) and establishes a prediction rule to classify new
samples (test set).
The quality of prediction of both microarray and
clinical data can depend on many factors, e.g. quality
of collected datasets, quantity of samples in datasets,
balance, relevance of used variables, etc. Ideally, vari-
ables should represent the changes caused by a dis-
ease. The quality of disease outcome predictor is de-
pendent on a machine learning method, on the pro-
cess of training and other effects. In literature, there
are many examples that do not evaluate microarray
experiments correctly (Dupuy and Simon, 2007).
Prediction accuracy of combination of microar-
ray and clinical data depends on complementarity of
these two data sources. If data sources or data mod-
els are complementary, i.e. they contain some non-
redundant information, combination of models leads
to increased prediction accuracy. Prediction accuracy
also depends on quality of models. In case the data
sources are redundant, pre-validation of microarray
and clinical data can assess quality of this data or the
models. The concept of pre-validation for microarray
108
Šilhavá J. and Smrž P. (2010).
IMPROVED DISEASE OUTCOME PREDICTION BASED ON MICROARRAY AND CLINICAL DATA COMBINATION AND PRE-VALIDATION.
In Proceedings of the First International Conference on Bioinformatics, pages 108-113
DOI: 10.5220/0002697601080113
Copyright
c
SciTePress

and clinical data is presented in (Tibshirani and Efron,
2002).
This paper describes a combination of logistic re-
gression for clinical data and BinomialBoosting for
microarray data. Then it describes its extension de-
signed for redundant sets of data. Microarray and
clinical data are combined at the levelof decision inte-
gration. The characters of logistic regression and Bi-
nomialBoosting models allow for their combination,
see Section 2. BinomialBoosting (Buhlmann and
Hothorn, 2007) enables use of logistic regresion with
high-dimensional data, which is impossible without
dimension reduction step and with high-dimensional
data. Logistic regression with high-dimensional data
can produce numerically unstable estimates and the
predicting model does not generalize well (Hosmer
and Lemeshow, 2000). In contrast to combined logis-
tic regression and BinomialBoosting models, the sec-
ond approach includes pre-validation of models built
with microarray and clinical data followed by weights
calculation. Weights set relevance of microarray and
clinical models for data combination.
The paper is organized as follows: Section 2 de-
scribes the combination of logistic regression and Bi-
nomialBoosting, then it describes its extension in-
cluding pre-validation. Simulations are performed
with several generated datasets in redundant and non-
redundant setting together with some tests applied to
two real benchmark datasets in Section 3. Some re-
lated work is shortly discussed in Section 4. Section 5
concludes this paper.
2 METHODS
2.1 Microarray and Clinical Data
Combination
Notation: Let Z be the n×q matrix with n samples and
q-dimensional clinical data. The response variable is
a n-dimensional vector Y. Then let X be other matrix
with microarray data. X is the n× p matrix containing
n samples and the expression values of p genes.
This approach consists of the two models: logis-
tic regression (LOG) and BinomialBoosting (BB), see
Figure 1. In a very brief description, BinomialBoost-
ing consists of the estimate initialization and then for
1 to M boosting iterations: (1) the negative gradient
vector is computed, (2) the negative gradient vector is
fitted by the componentwise linear least squares as the
base procedure and finally (3) the estimate and the co-
efficients are updated. The optimal number of boost-
ing iteration is the main tuning parameter which is
LOG
BB
X
η
+
inverse
logit
Y
Z
X
Z
η
Figure 1: The schematic drawing of microarray and clinical
data combination. Logistic regression and BinomialBoost-
ing models are trained just with training part of clinical and
microarray dataset.
determined with Akaike information criterion (AIC)
(Hothorn and Buhlmann, 2007).
Our approach combines microarray and clinical
data at the level of decision integration. This means
that separate models for microarray and clinical data
are trained and then the predictions of these models
(η
Z
and η
X
) are combined. The combination of the
outputs of these models is possible because the out-
puts of these models are linear and there are some
similar properties of logistic regression (Hosmer and
Lemeshow, 2000) and BinomialBoosting (Buhlmann
and Hothorn, 2007).
Similar properties of logistic regression and Binomi-
alBoosting:
• generalized linear models:
Y
i
= g(η
i
) , (1)
where g is a link function. η
i
is a linear model:
η
i
= β
0
+
k
X
j=1
β
j
Q
i, j
for i = 1, . . . , n , (2)
where β denotes coefficients, k and Q can be spec-
ified as: p and X for microarray data; q and Z for
clinical data.
• response variable Y
i
is considered binomial
(Bernoulli) random variable p
i
: Y
i
∼ binomial(p
i
, n).
Binomial response variables relate to logit function:
logit(p) = log
p
1−p
. Inverse logit is the link func-
tion g in logistic regression (1). In BinomialBoosting,
logit function is included in binomial loss function as
a population minimizer. BinomialBoosting with the
componentwise linear least squares as a base proce-
dure yields a fit of a linear logistic regression model
(Buhlmann and Hothorn, 2007).
This approach of combination of microarray and clin-
ical data can be described as follows, see Figure 1.
Microarray and clinical data are repeatedly split into
training and test sets via Monte Carlo cross-validation
(MCCV) procedure, see (Molinaro et al., 2005). Each
clinical training set is fitted to logistic regression
IMPROVED DISEASE OUTCOME PREDICTION BASED ON MICROARRAY AND CLINICAL DATA
COMBINATION AND PRE-VALIDATION
109
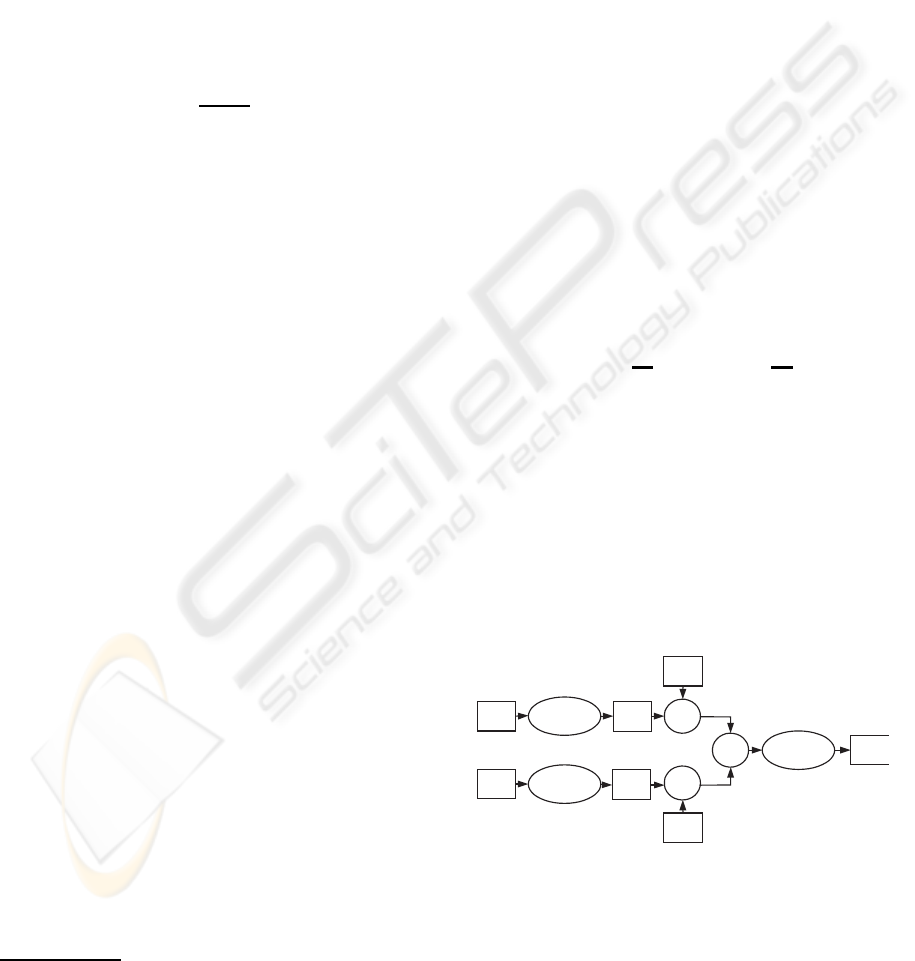
model. Then the linear prediction of each clinical test
set gives predictions η
Z
i
of the linear model (2) de-
noted for clinical data with the upper index Z. Each
microarray training set is fitted to the model using Bi-
nomialBoosting. Then the linear prediction of each
microarray test set gives predictions η
X
i
of the linear
model (2) denoted for microarray data with the upper
index X. According to the additivity rule that is valid
for linear models, we can sum the linear predictions:
η
i
= η
Z
i
+ η
X
i
. (3)
Then the logit inversion of η
i
gives a response:
Y
i
=
e
η
i
1+ e
η
i
. (4)
In the rest of this paper, this approach is denoted
as LOG/Z+BB/X
1
.
2.2 Microarray and Clinical Data
Pre-validation and Combination
This approach, in contrast to microarray and clini-
cal data combination, sets weights that determine rel-
evance of linear predictions for combination of mi-
croarray and clinical models, as shown in Figure 2.
This approach was designed for redundant datasets.
Weights are set based on pre-validation. The con-
cept of pre-validation for microarray data and clinical
variables is described in (Tibshirani and Efron, 2002).
This paper incorporates only points 1 through 5 com-
pared to our approach described in Section 2.3. Also,
we use different classifiers and leave-one-out cross-
validation (LOOCV), while (Tibshirani and Efron,
2002) uses k-fold CV.
2.3 Determination of Weights for
Models
We have K training samples in t-iteration of MCCV.
We use LOOCV for pre-validation and consequently
we determine weights. The weights are determined as
follows:
1. Set aside one sample of K training samples.
2. Build model with logistic regression (Binomial-
Boosting) for Z (X)
2
using only data from the
other K − 1 samples.
3. Predict linear response with built modelon left out
case.
1
In the rest of this paper, a slash in a title separates a
model and type of data.
2
Z denotes clinical data and X denotes microarray data.
4. Repeat steps 1–3 for each of the samples K to get
pre-validated predictors from Z and X.
5. Fit logistic regression model to pre-validated pre-
dictors from Z and X.
6. Compute weights w
i
(6), where i denotes Z or X.
7. Repeate steps 1-6 for randomized training data
obtained from MCCV.
8. Compute modus of weights ˆw
i
from w
i
for X and
Z.
In this approach, logistic regression is used
twice—in building model of Z and in building model
of pre-validated predictors from Z and X. Logistic
regression describes the relationship between one or
more variables and an outcome. Each of coefficients
describes the size of the contribution of each variable.
Large regression coefficient means that the variable
strongly influences the probability of that outcome.
The folowing equation for Z and X variable is derived
from (2):
η = β
0
+ β
Z
Q
Z
+ β
X
Q
X
. (5)
Then the weights are determined as follows:
w
Z
= abs
β
Z
β
0
, w
X
= abs
β
X
β
0
. (6)
In this approach, randomized training data ob-
tained from MCCV is used twice—in weights esti-
mation as described in Section 2.2 and in building
model of Z and X as described in Section 2.1. His-
togram of weights obtained from t-iteration of MCCV
is close to exponential distributions of probability
density function. In the case of exponential distribu-
tion, the modus is the value with the highest density.
In the rest of this paper, this approach is denoted
as pre-LOG/ Z+BB/X.
Z
s
w
~
X
s
w
~
+
inverse
logit
Y
LOG
BB
X
Z
Z
X
×
×
η
η
Figure 2: The schematic drawing of microarray and clinical
data pre-validation and combination. Logistic regression
and BinomialBoosting models are trained just with training
part of clinical and microarray dataset.
BIOINFORMATICS 2010 - International Conference on Bioinformatics
110

Table 1: Non-redundant datasets. AUCs from test datasets (including mean AUCs and standard deviations) evaluated over
100 MCCV iterations.
1. 2. 3. 4.
µ
Z
, µ
X
0, 0 1, 0.25 0.5, 0.5 1, 0.5
Method (no power) µ
Z
> µ
X
µ
Z
< µ
X
(strong p.)
LOG/Z+BB/X 0.53 ± 0.05 0.69 ± 0.04 0.73 ± 0.04 0.79 ± 0.04
LOG/Z 0.55 ± 0.05 0.65 ± 0.06 0.56 ± 0.05 0.65 ± 0.06
BB/X 0.51 ± 0.05 0.60 ± 0.05 0.72 ± 0.05 0.72 ± 0.05
Table 2: Redundant datasets. AUCs from test datasets (including mean AUCs and standard deviations) evaluated over 100
MCCV iterations.
1. 2. 3. 4.
µ
Z
, µ
X
0, 0 1, 0.25 0.5, 0.5 1, 0.5
Method (no power) µ
Z
> µ
X
µ
Z
< µ
X
(strong p.)
LOG/Z+BB/X 0.51 ± 0.05 0.94 ± 0.02 0.96 ± 0.02 0.98 ± 0.01
LOG/Z 0.49 ± 0.05 0.94 ± 0.02 0.78 ± 0.04 0.94 ± 0.02
BB/X 0.51 ± 0.05 0.71 ± 0.04 0.98 ± 0.01 0.98 ± 0.01
3 RESULTS
The focus of evaluation was to test LOG/Z+BB/X ap-
proach with non-redundant and redundant datasets.
Simulated data was used for this purpose. Then
we tested LOG/Z+BB/X and pre-LOG/Z+BB/X with
two real benchmark datasets. We performed experi-
ments in R environment
3
using packages ‘stats’ and
‘mboost’.
MCCV without replacement split the samples ran-
domly into a learning and test sets numerous times.
Large number of iterations lead to more stable results.
In our case, the whole procedure was repeated 100
times with learning set and test set ratio 4 : 1. We es-
timated the Area Under the ROC Curve (AUC) and
AUCs were averaged over 100 MCCV iterations.
3.1 Simulated Datasets
We tested LOG/Z+BB/X with non-redundant and re-
dundant datasets. We generated simulated datasets
through the use of R script available in (Boulesteix
et al., 2008). In case of redundant sets, microar-
ray and clinical variables are generated using exactly
the same model. Such variables discriminate classes
in the same way and giving redundant information.
In case of non-redundant sets, the observations are
assumed to form two distinct subgroups (Boulesteix
et al., 2008). Then we considered different predic-
tive powers for the clinical variables µ
Z
and differ-
ent predictive powers for the microarray variables
µ
X
. In present simulations, µ
Z
= 0 denotes no power,
µ
Z
= 0.5 moderate power and µ
Z
= 1 strong power for
3
www.r-project.org
Z. Similarly µ
X
= 0, 0.25, 0.5 for X. Difference in
µ
Z
and µ
X
ranges compensates for ranges of predictor
values for microarray and clinical variables.
The following Tables 1 and 2 display selected re-
sults of LOG/Z+BB/X for different predictive pow-
ers of Z and X. In case of non-redundant datasets,
LOG/Z+BB/X increases AUCs. LOG/Z+BB/X has a
good performance on redundant datasets as well.
3.2 Real Datasets
For evaluation, we used two benchmark breast can-
cer datasets (van’t Veer et al., 2002) and (Pittman
et al., 2004). The first one gives the expression levels
of 22483 genes for 78 breast cancer patients. Based
on existence of distant metastases, 34 of these sam-
ples are classified into the poor prognosis group, the
rest 44 samples belong to the the good prognosis
group. The used dataset is prepared as described in
(van’t Veer et al., 2002) and is included in R package
‘DENMARKLAB’ (Fridlyand and Yang, 2004). This
dataset includes 4348 resulting genes. Clinical vari-
ables are age, tumor grade, estrogen receptor status,
progesterone receptor status, tumor size and angioin-
vasion. The second one gives the expression levels
of 12625 genes for 158 breast cancer patients. Ac-
cording to recurrence of disease, 63 of these sam-
ples are classifed into the poor prognosis group, the
rest 95 samples belong to the good prognosis group.
The data was pre-processed using packages ‘gcrma’
and ‘genefilter’ to normalize and filter the data. The
genes that showed a low variability across all samples
were cleared out. The resulting dataset includes 8961
genes. Clinical variables are age, lymph node status,
IMPROVED DISEASE OUTCOME PREDICTION BASED ON MICROARRAY AND CLINICAL DATA
COMBINATION AND PRE-VALIDATION
111

Table 3: van’t Veer dataset. AUCs from test datasets (including mean AUCs and standard deviations) evaluated over 100
MCCV iterations. p denostes a number of microarray variables.
Method p = 50 p = 200 p = 500
LOG/Z+BB/X 0.79 ± 0.11 0.78 ± 0.11 0.79 ± 0.11
LOG/Z 0.82 ± 0.10 − −
BB/X 0.67 ± 0.13 0.65 ± 0.12 0.65 ± 0.11
pre-LOG/Z+BB/X 0.81 ± 0.10 0.82 ± 0.11 0.82 ± 0.10
Table 4: Pittman dataset. AUCs from test datasets (including mean AUCs and standard deviations) evaluated over 100
MCCV iterations. p denostes a number of microarray variables.
Method p = 50 p = 200 p = 500
LOG/Z+BB/X 0.79 ± 0.07 0.81 ± 0.08 0.82 ± 0.08
LOG/Z 0.67 ± 0.09 − −
BB/X 0.75 ± 0.08 0.77 ± 0.08 0.78 ± 0.08
pre-LOG/Z+BB/X 0.74 ± 0.08 0.76 ± 0.08 0.78 ± 0.08
estrogen receptor status, family history, tumor grade
and tumor size.
We perform tests for different numbers of vari-
ables (p = 50, 200, 500) in order to inspect efficiency
of both approaches. Variables are selected on the ba-
sis of the absolute value of the t-statistic using R pack-
age ‘st’.
Average AUCs and standard deviations over
100 MCCV iterations include Tables 3 and 4.
LOG/Z+BB/X perform with Pittman dataset well, see
Table 4. Pittman dataset approaches non-redundant
datasets and combination of microarray and clini-
cal data implicates outcome prediction improvement.
According to the results in Table 3, van’t Veer dataset
approaches redundant datasets. This finding coin-
cides with conclusion of (Gruvberger et al., 2003),
which points out a correlation of ER-alfa status in the
dataset generated by van’t Veer. LOG/Z+BB/X av-
erages linear predictions from microarray and clin-
ical models on redundant datasets. Compared to
LOG/Z+BB/X, pre-LOG/Z+BB/X improves AUCs
up to 4% in the case of real redundant dataset. Av-
erage AUC for pre-LOG/Z+ BB/X is 0.82.
4 RELATED WORK
The topic of combination or integration of microar-
ray and clinical data is not new. In literature,
there are more papers where authors describe vari-
ous ways of microarray and clinical data combination.
In principle, the results of designed approaches are
hard to compare because new approaches are evalu-
ated with different datasets and measures. (Gevaert
et al., 2006) evaluate with using AUC as our pa-
per. They integrate microarray data and clinical vari-
ables with Bayesian networks in three ways: full
integration, decision integration and partial integra-
tion. Their Bayesian decision integration approach
combines data at the same level as our method and
achieves average AUC 0.79 with van’t Veer dataset.
In order to compare pre-LOG/Z+BB/X with the ap-
proach proposed in (Boulesteix et al., 2008), we have
performed our simulations also in terms of mean er-
ror rates. Our approach provides results 2% better
on average on the van’t Veer dataset. In the case of
Pittman dataset, LOG/Z+BB/X has results 5% bet-
ter than the approach proposed in (Boulesteix et al.,
2008). The method described in this article employs
pre-validation principle with PLS dimension reduc-
tion. Random forests are then applied with new com-
ponents and the clinical variables as predictors. (Eden
et al., 2004) reproduce van’t Veer classifier for mi-
croarray predictors and apply an artificial neural net-
work (ANN) algorithm to clinical predictors. Their
approach achievesAUC 0.79 with all samples of van’t
Veer dataset and with LOOCV. Then this approach
achieves AUC 0.85 with only ER positive samples
of van’t Veer dataset. (Ma and Huang, 2007) pro-
pose Cov-TGDR method for combining different type
of covariates in disease prediction. They use van’t
Veer dataset and achieveprediction error 0.227. How-
ever, they perform feature selection based on the bi-
nary outcome with training and test data which is not
correct (pre-processing step 4 and 5 in this article).
Other examples of methods that combine microarray
and clinical data are (Fernandez-Teijeiro et al., 2004;
Pittman et al., 2004), but these authors evaluate sur-
vival times. (Fernandez-Teijeiro et al., 2004) build
predictive model with combination of clinical vari-
ables and a small number of selected genes. (Pittman
et al., 2004) combine metagenes with clinical risk fac-
tors to improve prediction.
BIOINFORMATICS 2010 - International Conference on Bioinformatics
112

5 CONCLUSIONS
This article deals with outcome prediction of com-
bined models. We combined microarray and clini-
cal data. We described LOG/Z+BB/X approach and
its extension pre-LOG/Z+BB/X designed for redun-
dant datasets. In contrast to LOG/Z+BB/X, pre-
LOG/Z+BB/X includes pre-validationof models built
with microarray and clinical data followed by weights
calculation. Weights set relevance of microarray and
clinical models for data combination. We evalu-
ated LOG/Z+BB/X with non-redundant and redun-
dant simulated datasets for different predictive powers
of microarray and clinical variables. LOG/Z+BB/X
increases AUCs on non-redundant simulated datasets
and it does not decrease AUCs on redundant sim-
ulated datasets. Then we evaluated LOG/Z+BB/X
and pre-LOG/Z+BB/X on two benchmark breast can-
cer datasets. LOG/Z+BB/X increases AUCs on
Pittman dataset. Compared to LOG/Z+BB/X, pre-
LOG/Z+BB/X improves outcome of the prediction
up to 4% in the case of van’t Veer dataset. Aver-
age AUC for pre-LOG/Z+BB/X is 0.82. In conclu-
sion, LOG/Z+BB/X performs with combined mod-
els well—both with non-redundant data and redun-
dant data. When this approach does not perform well,
it is possible to apply pre-LOG/Z+BB/X approach
or evaluate the quality of data or models separately.
Plans to the future include incorporation of other
data sources into combination and deriving biomark-
ers significantly involved in outcome prediction.
ACKNOWLEDGEMENTS
This work was partly supported by the Czech Min-
istry of Education research grants 2B06052 and
MSM0021630528. We thank Petr Holub and the re-
viewers for their constructive comments.
REFERENCES
Boulesteix, A. L., Porzelius, C., and Daumer, M. (2008).
Microarray-based classification and clinical predic-
tors: on combined classifiers and additional predic-
tive value. Bioinformatics 24, 1698-1706.
Buhlmann, P. and Hothorn, T. (2007). Boosting Algorithms:
Regularization, Prediction and Model Fitting. Statist.
Sci. 22, 477-505.
Dupuy, A. and Simon, R. M. (2007). Critical Review of
Published Microarray Studies for Cancer Outcome
and Guidelines on Statistical Analysis and Reporting.
Journal of the National Cancer Institute 99 (2), 147-
157.
Eden, P., Ritz, C., and Rose, C. (2004). Good Old clinical
markers have similar power in breast cancer progno-
sis as microarray gene expression profilers. Eur. J.
Cancer 40 (12), 1803-1806.
Fernandez-Teijeiro, A., Betensky, R. A., Sturla, L. M., Kim,
J. Y., Tamayo, P., and Pomeroy, S. L. (2004). Combin-
ing gene expression profiles and clinical parameters
for risk stratification in medulloblastomas. J Clin On-
col. 22 (6), 994-998.
Fridlyand, J. and Yang, J. Y. H. (2004). DENMARK-
LAB R package. Advanced microarray data analysis:
Class discovery and class prediction. Available at
http://genome.cbs.dtu.dk/courses/norfa2004/Extras/.
Gajdos, C., Tartter, P. I., and Bleiweiss, I. (1999). Lym-
phatic Invasion, Tumor Size, and Age Are Indepen-
dent Predictors of Axillary Lymph Node Metastases in
Women With T1 Breast Cancers. Ann Surg. 230 (5),
692-696.
Gevaert, O., Smet, F. D., and Timmerman, D. (2006). Pre-
dicting the prognosis of breast cancer by integrating
clinical and microarray data with Bayesian networks.
Bioinformatics 22 (14), 184-190.
Gruvberger, S. K., Ringner, M., and Eden, P. (2003). Ex-
pression profiling to predict outcome in breast cancer:
the influence of sample selection. Breast Cancer Res.
5(1), 23-26.
Hosmer, D. W. and Lemeshow, S. (2000). Applied Logistic
Regression. Wiley, New York, 2nd edition.
Hothorn, T. and Buhlmann, P. (2007). mboost: Model-
Based Boosting. R package version 0.5-8. Bioinfor-
matics, Available at http://CRAN.R-project.org/.
Klijn, J. G. M., Wang, Y., Atkins, D., and Foekens, J. A.
(2005). Prediction of cancer outcome with microar-
rays. Lancet. 365 (9472), 1685-1685.
Ma, S. and Huang, J. (2007). Combining Clinical and Ge-
nomic Covariates via Cov-TGDR. Cancer Inform. 3,
371-378.
Michiels, S., Koscielny, S., and Hill, C. (2005). Prediction
of cancer outcome with microarrays: a multiple ran-
dom validation strategy. Lancet. 365 (9458), 488-492.
Molinaro, A., Simon, R., and Pfeiffer, R. M. (2005). Pre-
diction error estimation: a comparison of resampling
methods. Bioinformatics 21(15), 3301-3307.
Pittman, J., Huang, E., and Dressman, H. (2004). Inte-
grated modeling of clinical and gene expression in-
formation for personalized prediction of disease out-
comes. Proc.Natl.Acad.Sci. 101(22), 8431-8436.
Tibshirani, R. and Efron, B. (2002). Pre-validation and in-
ference in microarrays. Statistical applications in ge-
netics and molecular biology 1, 1.
van’t Veer, L. J., Dai, H., and van de Vijver, M. J. (2002).
Gene expression profiling predicts clinical outcome of
breast cancer. Nature 415, 530-536.
IMPROVED DISEASE OUTCOME PREDICTION BASED ON MICROARRAY AND CLINICAL DATA
COMBINATION AND PRE-VALIDATION
113
