
CO-EVOLUTION IN HIV ENZYMES
P. Boba, P. Weil, F. Hoffgaard and K. Hamacher
Bioinformatics & Theoretical Biology Group, Technische Universit
¨
at Darmstadt, Germany
Keywords:
Sequence analysis, Human immunodeficiency virus, HIV, Co-evolution, Mutual information, Data mining,
Machine learning.
Abstract:
Proteins as molecular phenotypes need to maintain their stability, fold, and the functionality throughout their
individual and collective evolution. Such important properties are maintained by a selective pressure that
reveals itself in sequence data sets. Small adaptive changes are usually possible, but in general the conservation
of structure and function implies the co-evolution of amino acids within the molecule. We analyze two most
important enzymes in the progression of viral infection by the human immunodeficiency virus (HIV) – namely
the reverse transcriptase and the protease – under an information theoretical framework to derive insight into
the selective pressure acting locally and globally on the enzymes. To this end we computed mutual information
inside the proteins and between the proteins for some 40,000 sequences. We discuss the results of intra- and
inter-protein co-evolution of residues in these enzymes and finally annotate important structural-evolutionary
correlations. In particular we focus on the reverse transcriptase and a small signal indicating a potential co-
evolution between the protease and the reverse transcriptase. We convinced ourselves that our sampling is
sufficiently large and that no normalization schemes needs to be applied. We conclude with a short outlook
into potential implications for drug resistance development.
1 INTRODUCTION
The acquired immunodeficiency syndrome (AIDS)
is induced by the human immunodeficiency virus
(HIV). Its viral replication cycle depends on the virus
own protease and several other enzymes such as the
reverse transcriptase. Currently the anti-HIV drugs
target these two enzymes to prevent the maturation of
new virions (Tsygankov, 2009; Wlodawer and Erick-
son, 1993).
Neutral evolution and drug resistance develop-
ment have been under investigation for a long time:
1) the high mutation rate of HIV makes the virus
an interesting evolutionary object in itself as it per-
forms a large-scale mutagenesis study (Perelson et al.,
1996); 2) a deeper knowledge of potential evolution-
ary barriers might lead to new therapeutics besides the
HAART-procedure (Richman et al., 2009).
The theoretical understanding of the viral evolu-
tion has greatly improved over the recent years (Rong
et al., 2007; Chen and Lee, 2006; Trylska et al., 2007;
Hamacher and McCammon, 2006), even the biophys-
ical annotation based on in silico models of the molec-
ular dynamics is under way (Hamacher, 2008).
At the same time the wealth of information on
HIV - in particular the large data sets of sequences
- prompt for a deeper analysis on the sequence level
alone. Here we leverage an available data set of
45,161 mutant sequences of the HIV-1 protease (PR)
and reverse transcriptase (RT).
2 MATERIALS AND METHODS
2.1 Sequence Data
The 45,161 positive selection mutant sequences have
been collected by the Lee lab (Pan et al., 2007; Chen
et al., 2004) and were made available on the net. The
data set contains the genomic, nucleotide sequences
from treated and untreated patients under various drug
regimes. The individual entries are, however, not an-
notated by the drug treatment regime of the particu-
lar patient. We therefore find in this data set the di-
verse evolutionary dynamics, including effects such
as neutral drift, drug resistance development, and
other selective pressures on the two enzymes. Wher-
ever a codon could not be mapped unequivocally to
an amino acid we used a wild-card character, treating
these cases similar to gaps.
For a comparison on the quality and potential
39
Boba P., Weil P., Hoffgaard F. and Hamacher K. (2010).
CO-EVOLUTION IN HIV ENZYMES.
In Proceedings of the First International Conference on Bioinformatics, pages 39-47
DOI: 10.5220/0002731800390047
Copyright
c
SciTePress
finite-size effects we created also sequence align-
ments with CLUSTALW (Thompson et al., 1994;
Higgins and Sharp, 1988) and standard parameters on
1. BLAST (Altschul et al., 1990) hits on a sequence
of viral ion channel Kcv, and 2. ribosomal proteins
from bacterial genomes extracted by Pfam Hidden-
Markov-Models (Finn et al., 2008).
2.2 Information Theoretical Measures
on (Co-)Evolution
The evolution of an amino acid at a position i means
a change in the symbols S
i
over time within a set of
acceptable values S . One way to quantify the infor-
mation content of such collections of symbols is the
Shannon entropy (Shannon, 1951)
H
i
:= −
∑
S
i
∈S
p(S
i
) · log
2
p(S
i
)
(1)
where p(S
i
) is the probability of the occurrence of the
symbol S
i
within the empirical or theoretical data set
under investigation. For empirical data sets one usu-
ally sets this probability to the frequency of the sym-
bol within the data set. Positions (in e.g. a sequence
alignment) with high entropy are then amino acids
with high variability during evolutionary times. Our
choice of S comprised the 20 standard amino acids
and the above mentioned wild-card character.
The correlated change in the amino acid com-
position within a molecule or between molecules is
now based on empirical found two-point probabilities
p(S
i
,S
j
) for the co-evolution of positions i and j. We
can define the Mutual Information (MI) between these
positions as a relative entropy as follows (Lund et al.,
2005):
MI
i, j
:=
∑
S
i
,S
j
∈S
p(S
i
,S
j
) · log
2
p(S
i
,S
j
)
p(S
i
) · p(S
j
)
= H
i, j
− H
i
− H
j
(2)
The value of the MI gives the amount of informa-
tion that one position i conveys about the other po-
sition j. The MI can be derived from the Kullback-
Leibler divergence as a relative entropy, which has
- besides sequence based approaches - also attracted
attention as a measure in in silico drug design and
molecular biophysics (Hamacher, 2007).
In addition we applied two normalization proce-
dures to the MI in the following form, of which one
was suggested earlier (Gloor et al., 2005) to account
for potential sampling artefacts:
MI
i, j
(2)
:= MI
i, j
/H
i, j
MI
i, j
(1,1)
:= MI
i, j
/(H
i
· H
j
) (3)
0 100 200 300 400 500 600 700
0.0
0.5
1.0
1.5
N
median MI
KCV
HIV1−Pr
S20
S6
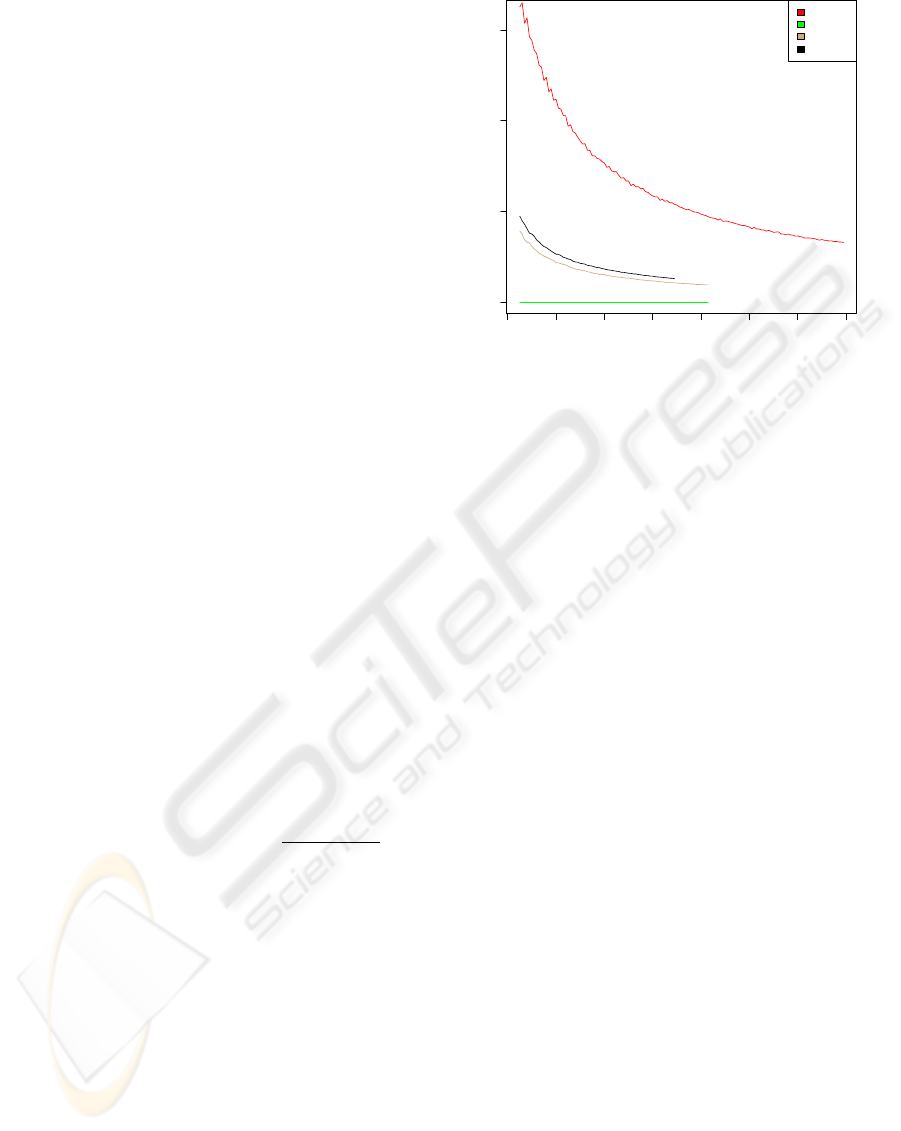
Figure 1: The median of the Mutual Information according
to equation 2 for sequence homologs of the ion channel Kcv
of plant viruses, bacterial ribosomal proteins S20 and S6,
and the HIV-1 protease. We display the mutual information
as a function of the number of sequences N included in the
computation.
These normalizations are principally necessary to
cope with finite data sets and potential absolute con-
servation of individual positions.
3 RESULTS
3.1 Finite Size Effects
To estimate size effects due to finite data we applied
an established protocol: in figure 1 we show the MI as
computed from randomized alignments of the listed
molecules. The randomization was performed inde-
pendently in each column by shuffling the characters.
A perfect randomized sample would provide for an
independence between columns i and j, and thus to
a vanishing two-point-distribution function p(S
i
,S
j
).
This leads in equation 2 also to a vanishing MI
i, j
=0.
This allows to achieve an understanding of the statis-
tical significance of a data set.
Clearly the data set for the viral enzymes under
consideration is sufficient in comparison to the se-
quence collections for Kcv and ribosomal proteins.
We analyzed this further and found the reason for this
in the high gap content [data not shown] of the non-
viral proteins chosen for comparison. The sequences
for HIV-1 PR, on the other hand, do not contain any
gap character at all, while the gap/wild-card character
content for the RT is also negligible.
BIOINFORMATICS 2010 - International Conference on Bioinformatics
40
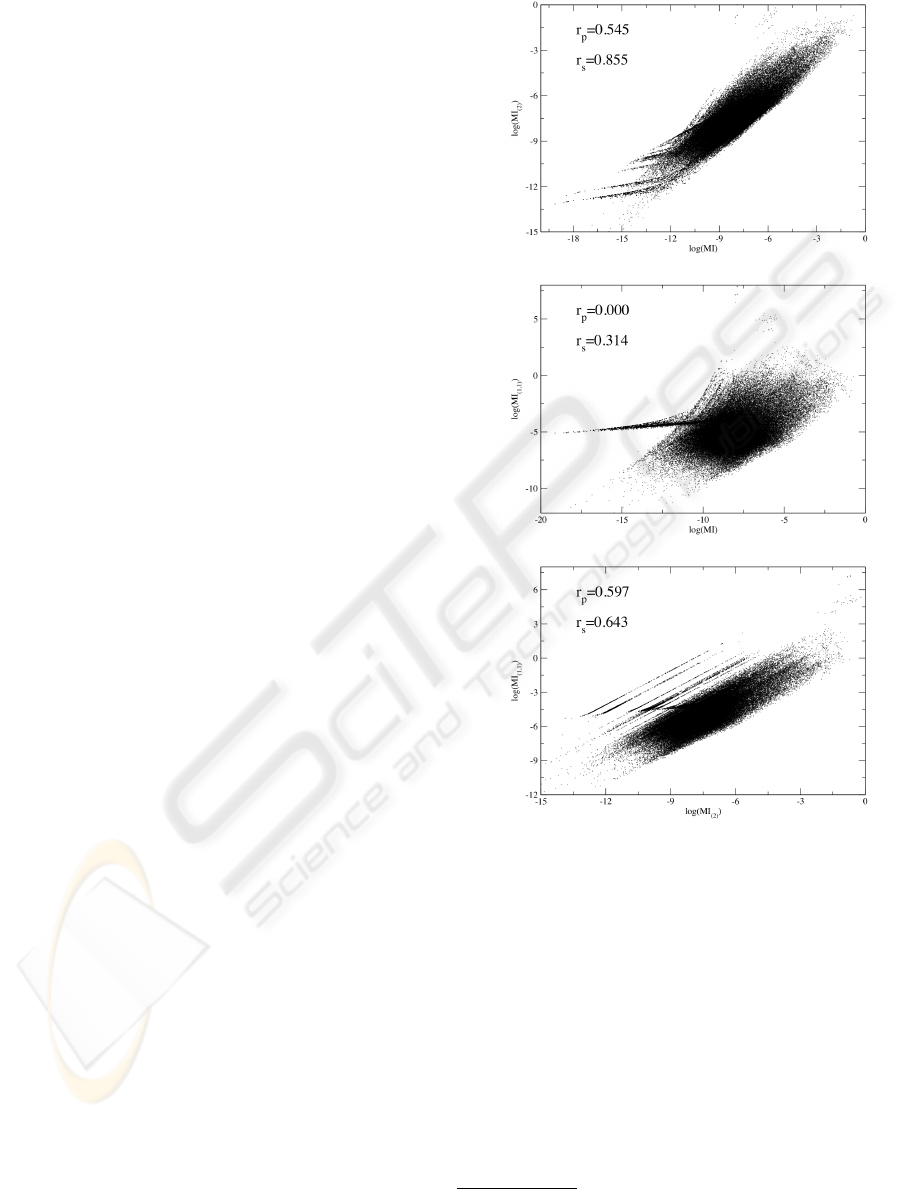
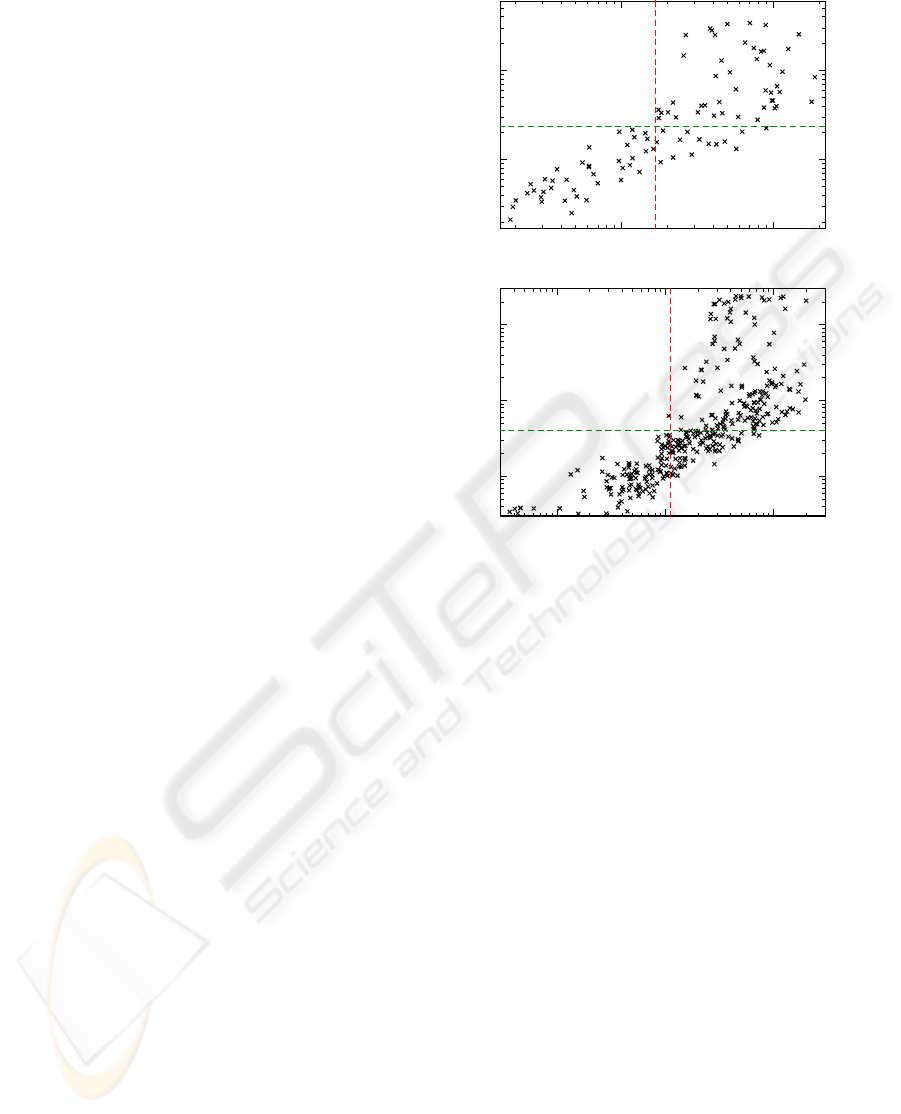
3.2 Effect of Normalization
We applied the two variants of the normalization pro-
cedure to the sequences of HIV-1 PR and RT. In fig-
ure 2 we show the change of the computed correlation
measures under those normalization procedures. We
applied a non-linear correlation measure - namely the
Spearman ranking coefficient - that is superior in re-
gard to the insight one gains into the relations between
the two data sets.
We note in passing that the results for the two nor-
malized MI variants MI
(2)
and MI
(1,1)
showed similar
distributions.
We draw three conclusions from the observations
in sections 3.1 and 3.2:
• effects to the finite-size of our data set are negli-
gible
• MI
(2)
was shown (Gloor et al., 2005) to take finite-
size effects most reliable into account
• the high Spearman correlation between MI
(2)
and
MI indicate an equivalence
We therefore will in the subsequent parts of the dis-
cussion in this manuscript focus solely on the MI as
there is no additional gain in using any normalization
in this particular case.
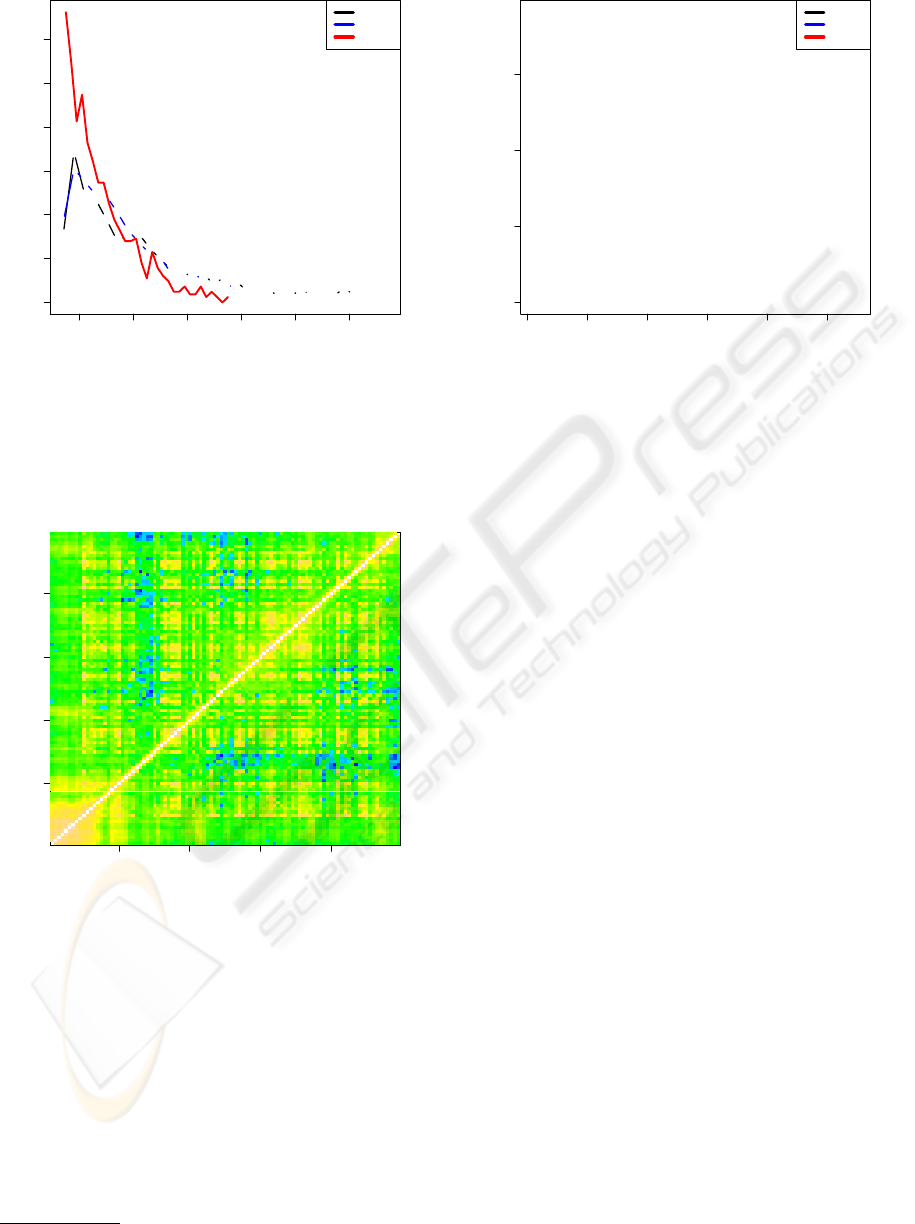
3.3 Comparing Intra- and Inter-Protein
Co-Evolution of Residues
In figure 3 we show distributions of the naked MI-
values from our study on both, the HIV-1 PR and the
HIV-1 RT, as well as the inter-MI for a potential co-
evolution of residues in these enzymes.
We observe similarity of MI results for the intra-
co-evolution within the individual, isolated enzymes.
Obviously the evolutionary dynamics gave rise to the
same overall “mutual information picture”.
The dissimilarity of the MI-distributions for the
RT/PR and the one for the inter-molecular MI comes
as no surprise: within a molecule the evolutionary
pressure on the co-evolving dynamics of amino acids
can be regarded as quite different in the evolution-
ary dynamics between residues in different molecules,
despite potential protein-protein-interactions or other
implicit interdependencies resulting from cell biolog-
ical effects or drug combinations.
Although the RT consists of four domains –
namely the finger, palm, thumb, connection domains
– the potential for co-evolution between sites dis-
tributed over the four domains runs approximately in
parallel to the scenario of the protease, both of which
- in turn - are constructed as a molecular phenotype in
form of homodimers.
a)
b)
c)
Figure 2: Scatter plots of the natural logarithm of the MI
values, comparing normalization procedures of eqs. 3.
a) MI
i, j
and MI
i, j
(2)
; b) MI
i, j
and MI
i, j
(1,1)
; c) MI
i, j
(1,1)
and
MI
i, j
(2)
. In each figure we give the Pearson correlation r
p
and the Spearman ranking coefficient r
s
(W.H. Press et al,
1995) between the data points. Note that MI values smaller
than 10
−8
were omitted for numerical reasons.
In figure 4 we show a graph for the MI of the HIV-
1 PR that indicates structural as well as dynamical
features as discussed in (Hamacher, 2008). We omit a
picture for the RT as the molecule is too large to dis-
play single MI entries on single-pixel basis. The raw
data, however, is available from our web-site (Boba
and Hamacher, 2009) for future analysis
1
.
1
Work is underway to construct a software-package to
visualize such voluminous matrices as for the RT (Schreck
CO-EVOLUTION IN HIV ENZYMES
41
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
● ●
●
●
●
●
● ●
−5 −4 −3 −2 −1 0
0.0 0.2 0.4 0.6 0.8 1.0 1.2
log(MI)
Density
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
PR
RT
PR−RT
Figure 3: Comparison of the MI-values for the intra-protein
co-evolution within the HIV-1 Protease (black) and the
HIV-1 Reverse Transcriptase (blue). We compare to the
inter-MI for the co-evolution between residues of the HIV-1
Protease on the one hand and the HIV-1 Reverse Transcrip-
tase on the other (red).
20 40 60 80
20 40 60 80
ID
ID
Figure 4: The logarithm of the MI within the HIV-1 PR for
all pairings of residue numbers. Clearly we reproduce fea-
tures already found in (Hamacher, 2008) and have therefore
verified our analysis protocol.
To analyze our MI results further and to overlay
these with structural knowledge, we went on with
a spectral decomposition of the MI matrices for the
HIV-1 PR and HIV-1 RT. For the inter-MI values,
that would indicate potential co-evolution between
residues of different molecules, the MI matrix is,
however, non-quadratic as the protein lengths are in
general different. We therefore applied a singular
et al., 2009).
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
0 10 20 30 40 50
0.0 0.5 1.0 1.5
k
λλ
k
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●
●
● ●
● ●
●
●
PR
RT
PR−RT
Figure 5: The leading eigenvalues λ
k
of a spectral decom-
position of the MI-matrices. In black we show the ones
for the (99 × 99) MI-matrix of HIV-1 PR, in blue for the
(348× 348) MI-matrix of HIV-1 RT (we restricted our anal-
ysis to the first 348 residues to cope with bad sequence res-
olution towards the C-terminus of the RT in the underly-
ing data set). The red points give the singular values of the
singular value decomposition (W.H. Press et al, 1995) of
the inter-co-evolution matrix, which reflects MI-values be-
tween residues in the PR on the one hand and the RT on
the other. The fast decay of the λ
k
justifies a reconstruction
of the respective MI-matrices by just a few, even only one
eigenvector.
value decomposition (W.H. Press et al, 1995) to ob-
tain a pseudo-spectral decomposition with respect to
the singular values of the inter-MI matrix. The fast
decay of the eigenvalues/singular values as shown in
figure 5 indicates that a reconstruction of the whole
MI matrices can be achieved by just a few eigenvec-
tors, thus this small set of eigenvectors contains most,
if not all, of the mutual information.
If we now overlay these eigenvectors onto the
structures of the molecules, we can immediately con-
nect structural and evolutionary information. This is
done in figures 6 and 7.
Figures 6 and 7 both show high mutual informa-
tion for secondary structure elements. In particular
the β-sheet in the PR needs to be maintained as a
structural basis of the fold of this protein. This is
achieved by co-evolution of the residues within this
element. In the RT the β-sheet close to the reactive
center, as well as the α-helices forming the “finger”
of the RT are structurally maintained by co-evolving
the residues without giving them the freedom to inde-
pendently mutate.
In figure 7 we show in addition relevant residues
as small spheres. The colors indicate: yellow=three
catalytic aspartic acids; green=residues that enhance
BIOINFORMATICS 2010 - International Conference on Bioinformatics
42
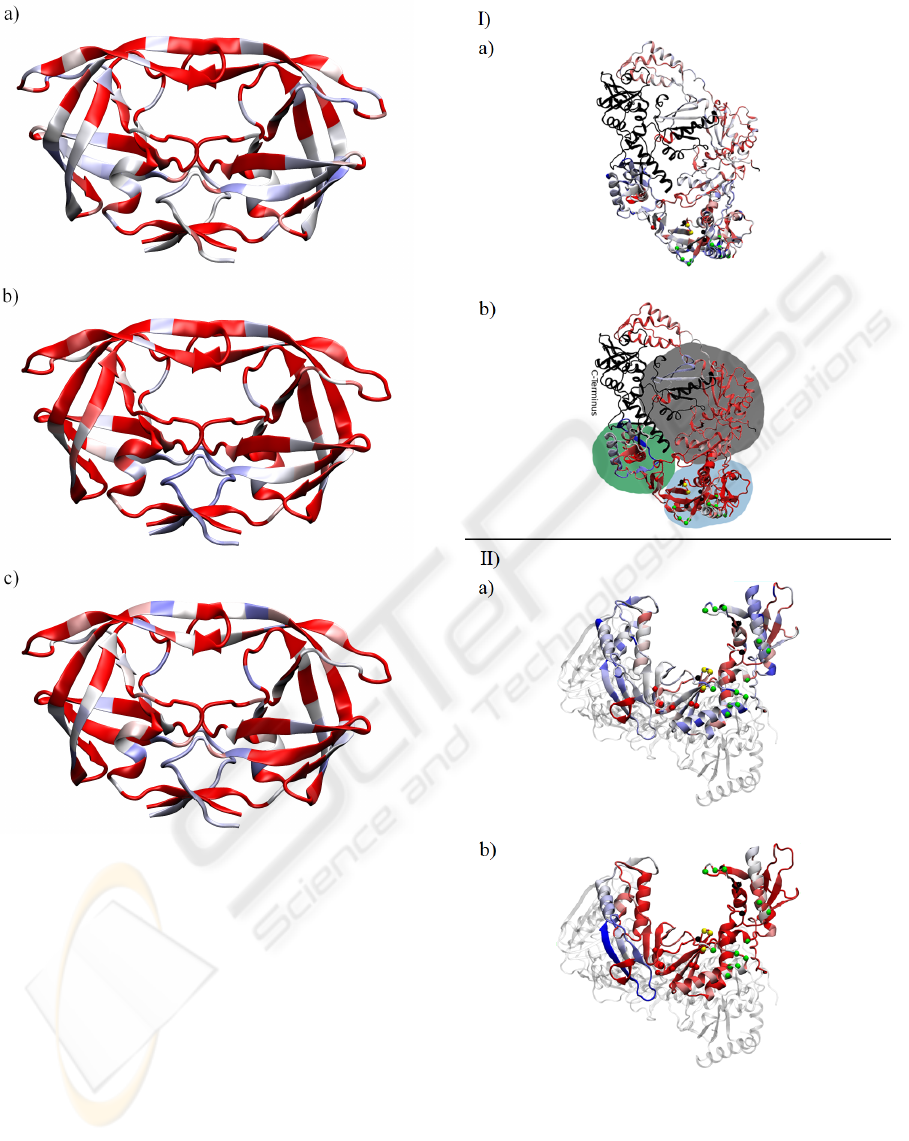
Figure 6: a) sequence entropy of the HIV-1 PR as in eq. 1;
b) absolute values of the entries of the 1
st
eigenvector for
the MI matrix of HIV-1 PR; c) absolute values of the en-
tries of the 2
nd
eigenvector. We rescaled all values so that
blue=maximum value, red=minimum value.
the excision reaction; red=non-nucleoside inhibitor
binding pocket; black=residues involved in NRTI-
resistance. This mapping was done in accordance
with previous work (Sarafianos et al., 2004).
We note in passing that the level of conservation
needs to be taken into account: an absolutely con-
served position shows a MI of zero, always, as the
knowledge about the identity of a residue here does
not convey any information on any other position. We
therefore decided to also display the sequence entropy
of equation 1 as a measure of local sequence conser-
vation in the figures 6 and 7. We return to this issue
Figure 7: I) a) sequence entropy of the HIV-1 RT as in eq. 1;
b) absolute values of the entries of the 1
st
eigenvector for
the MI of HIV-1 RT. II) viewed from orthogonal projection
& rescaled as in fig. 6. The black part is the C-terminus for
which we had only insufficient statistics; we omitted it from
our analysis. The small spheres indicate functional sites as
discussed in the text. Domains are indicated as follows:
light blue=”fingers”, green=”thumb”, gray=”palm”.
CO-EVOLUTION IN HIV ENZYMES
43
in the disucssion section.
Furthermore one can see the “correlation” of low
sequence entropy and therefore the necessarily low
mutual information in figure 8. In this figure we mo-
tivate the classification of an amino acid by its two
evolutionary/co-evolutionary measures, that is the se-
quence variability as expressed by the entropy of eq. 1
and the contribution to mutual information correlation
expressed by the respective entry in the leading eigen-
vector.
Class I (low sequence entropy, high MI) must be
empty. Classes II and IV are the important ones for
evolution: amino acids found in class IV are subject
to extensive selective pressure to maintain their iden-
tity (low sequence entropy, thus small sequence vari-
ability). Evolution acts here locally to force sequence
stabilization. Amino Acids in class II on the other
hand can vary extensively (large sequence entropy),
but at the same time convey information about other
amino acids, thus show correlation with other sites
within the protein. Accordingly the high MI reflects a
selective pressure to maintain not a particular amino
acid character, but instead to maintain some “interac-
tion”. This “interaction” might be a physical, direct
interaction such as steric repulsion or charge inter-
actions, but could also reflect, e.g. folding properties
of a monomer or recognition capabilities in protein-
protein-binding mechanisms. One might hypothesize
about the origin of this correlation or “interaction”,
but a high MI indicates always a selective pressure to
connect residues.
Class III on the other hand consists of those amino
acids, which are highly variable (high sequence en-
tropy), but at the same time show low dependence on
other sites within the protein, and thus low connection
via MI to these other positions.
3.4 Co-Evolution between Residues in
the PR and the RT?
In figure 3 we have seen that the inter-MI is some
two orders of magnitude smaller than the overall MI
of the intra-MI. Combining this insight with the typ-
ical values found in MI studies as shown in figure 1
we nevertheless find a basal co-evolution between the
two enzymes under investigation.
Although such a co-evolution is counter-intuitive
at first sight, their might be some small or even un-
known interdependencies between the two molecules.
For example one effect might be due to the packing of
RNA in the viral capsid and the genes coding for the
enzymes are in close vicinity of the RNA. Such pack-
ing is highly susceptible to local charges and balances
thereof - probably leading to long-range correlations
a)
0.1 1
Sequence Entropy [bit]
0.01
0.1
|entry of first eigenvector of MI|
I
III
II
IV
b)
0.01 0.1 1
Sequence Entropy [bit]
0.001
0.01
0.1
|entry of first eigenvector of MI|
I
III
II
IV
Figure 8: a) The correlation of MI contributions as found
in the first eigenvector of the MI-matrix of PR vs. the re-
spective sequence entropy of the amino aids in the PR. We
applied an intuition driven classification scheme to decom-
pose the results into four classes, numbered by Roman let-
ters and illustrated by the red and green line. b) same as a),
but for the RT.
along the genomic sequence. Additional potential ef-
fects are discussed in the discussion section 4.
In figure 9 we show our results for the pseudo-
spectral reconstruction of the inter-MI between the
PR and the RT.
4 DISCUSSION & SUMMARY
In this paper we have analyzed two of the most impor-
tant enzymes for the progression of viral infection by
the human immunodeficiency virus (HIV-1 protease
and the HIV-1 reverse transcriptase) in an information
theoretical setting to investigate evolutionary dynam-
ics and extract positions under exceptional selective
pressure.
A first insight is possible by looking solely at the
sequence variability, which reveals selective pressure
to maintain local properties within the molecule - lo-
cal is meant here in the sense of an individual posi-
tion. Sites of enzymatic action are prone examples of
BIOINFORMATICS 2010 - International Conference on Bioinformatics
44
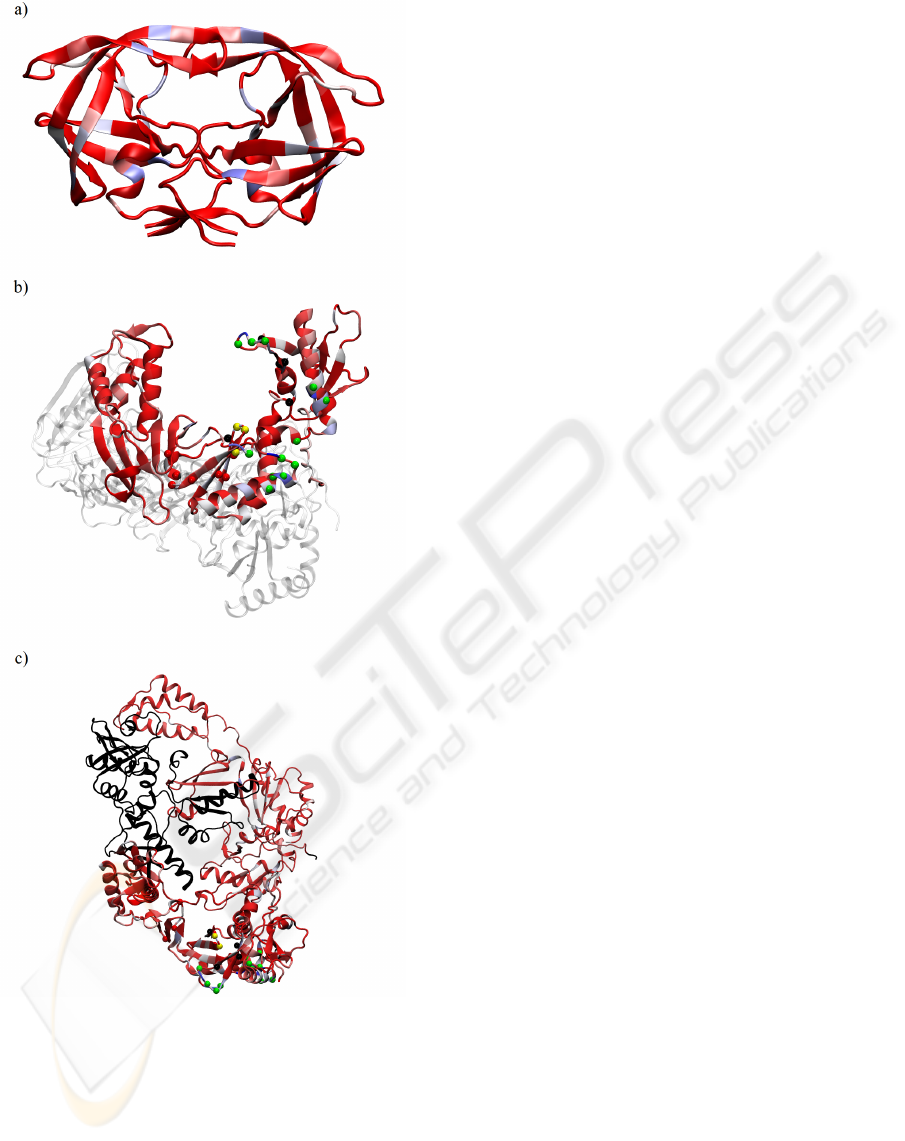
Figure 9: Absolute values of the entries of the 1
st
left-
and right-singular vector for a) the HIV-1 PR and b)+c)
for the HIV-1 RT. Again we rescaled all values so that
blue=maximum value, red=minimum value, and show the
non-analyzed parts of the RT in black.
such findings.
Nevertheless molecular evolution provides for an
extended selective pressure, which we label as non-
local as it involves several amino acids at the same
time. Despite individual amino acids being variable,
pairs of residues are connected or correlated. This is
revealed by the mutual information they carry.
We have shown that our sampling statistics is suf-
ficient and a standard normalization procedure usu-
ally applied is not necessary in our case - due to large
sample size and absence of gaps in aligned sequences.
A particularly interesting result is the high se-
quences variability in the β-sheets of the PR, as shown
in figure 6 a). At the same time we find these residues
also to be relevant for the high MI (parts b & c of the
same figure). This was recently discussed and anno-
tated in a biophysical simulation setting (Hamacher,
2008).
At the same time, we find one residue (I54 in the
wild-type) in the flaps to be highly variable and well
correlated to other parts of the PR, see the blue residue
in the upper strand of the β-sheet forming the flaps in
figure 6.
Interestingly in figure 6 the dimerization interface
of the PR in the lower part of the molecule shows
over a larger range high sequence variability as well
as large contributions to the mutual information. This
indicates HIV-1’s ability to vary the composition of
the binding interface to dimerize the PR-monomers to
become the PR-homodimer. Obviously maintaining
recognition capabilities for binding is of paramount
importance for the virus, revealing itself in the high
MI.
The implications of relating sequence variabil-
ity and mutual information can be seen in figure 8.
An intuitive classification scheme can be justified on
grounds of selective pressure induced by the ongoing
evolution of these molecular phenotypes and divided
in accordance with this classification procedure.
In table 1 we extracted the most pronounced
residues under these classification schemes - the ones
that correspond the most to the three existing classes.
To this end we have chosen visually those residues
most distant from the intersection of the red and green
lines in figure 8.
We find in table 1 the L10 and M46 for the pro-
tease to be of class II. Correlated mutations in these
positions are known to reduce binding of well-known
protease inhibitors, such as JE-2147 by an order of
magnitude or even more (Yoshimura et al., 1999;
Reiling et al., 2002). This makes the acquisition of
mutations relatively easy: these amino acids are not
to be preserved, they only need to maintain “interac-
tions” or correlations, thus opening the path to change
the sequence locally in a correlated fashion to reduce
drug efficiency while maintaining the structure, func-
tion, and thus the infectious outcome of the protease.
In class IV we found some of the amino acids
building the flaps of the PR (res. no. 52-58 are usu-
CO-EVOLUTION IN HIV ENZYMES
45
HIV-1 PR HIV-1 RT
I II III IV
63 41 49
10 69 78
71 57 29
12 60 56
none 20 70 28
7 61 27
90 16 86
82 39 52
46 67 98
54 92 51
I II III IV
334 102 349
335 49 348
329 108 347
333 106 344
none 339 249 346
338 48 343
324 165 345
322 100 342
326 90 341
311 4 152
Table 1: The most pronounced members of the classes as
introduced in figure 8 for both enzymes. For class I no
points exist that fulfill the particular requirement. The enu-
meration is in accordance with p66 monomer of the RT
dimer. We used the numbering convention of (Prajapati
et al., 2009; Sarafianos et al., 2004) for RT.
ally labeled to be part of the flaps). As is known
from extensive simulations (Perryman et al., 2006)
the flaps need to be most flexible to embrace the sub-
strate of the PR. This - as indicated by our findings - is
achieved evolutionary to strictly conserve the overall
sequence composition of the flaps.
For the RT we find in table 1 the class III very in-
teresting: residues of the binding pocket for the non-
nucleoside inhibitors are to be found here. Class III
contains, however, those positions that vary a lot, but
do not show high correlation to other positions in the
molecule. This implies that the amino acids bind-
ing the inhibitor can more or less freely mutate, be-
cause they are not correlated to other positions and
thus there is not need for correlated mutations, which
turned out to be necessary for the resistance develop-
ment of the protease (see above).
We found some indications for a potential co-
evolution between the PR and the RT. We can think
of three reasons to this end:
The weak co-evolution between the proteins
might be – as speculated in the results section – in-
duced by implicit interactions of the coding genes
during packing of the viral RNA into the capsid. Ob-
viously charge distributions play a prominent role
during these events and that might correlate (slightly)
nucleotides and therefore also the coded amino acids.
It is, however, reasonable to assume this effect to be
distributed all over the proteins and not localized on
particular residues.
Another selective pressure on both proteins is
collectively induced by application of protease and
reverse transcriptase inhibitors at the same time or
in temporal proximity, as in e.g. HAART treatment
(Richman et al., 2009), combining both types of in-
hibitors, for example lopinavir, ritonavir, tenofovir
and emtricitabine. We note that we at least in the RT
structure no contribution from portions of the com-
plex that bind RT inhibitors can be observed, making
this explanation less likely.
And finally one cannot completely neglect the
possibility of functional protein-protein-interactions
between the RT and the PR. Although there are cur-
rently no indications to this effect and we doubt that
they exist, we mention this possibility for the sake of
completeness here.
ACKNOWLEDGEMENTS
KH gratefully acknowledges financial support by the
Fonds der Chemischen Industrie through the program
Sachkostenzuschuß f
¨
ur den Hochschullehrernach-
wuchs.
Molecular Structures were visualized by VMD
(Stone, 1998; Humphrey et al., 1996). VMD was de-
veloped by the Theoretical and Computational Bio-
physics Group in the Beckman Institute for Advanced
Science and Technology at the University of Illinois
at Urbana-Champaign.
REFERENCES
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and
Lipman, D. J. (1990). Basic local alignment search
tool. J Mol Biol, (3):403–410.
Boba, P. and Hamacher, K. (2009).
http://bioserver.bio.tu-darmstadt.de/HIV.
Chen, L. and Lee, C. (2006). Distinguishing HIV-1 drug
resistance, accessory, and viral fitness mutations using
conditional selection pressure analysis of treated ver-
sus untreated patient samples. Biology Direct, 1(1):14.
Chen, L., Perlina, A., and Lee, C. J. (2004). Positive Selec-
tion Detection in 40,000 Human Immunodeficiency
Virus (HIV) Type 1 Sequences Automatically Iden-
tifies Drug Resistance and Positive Fitness Mutations
in HIV Protease and Reverse Transcriptase. J. Virol.,
78(7):3722–3732.
Finn, R. D., Tate, J., Mistry, J., Coggill, P. C., Sammut,
S. J., Hotz, H.-R., Ceric, G., Forslund, K., Eddy, S. R.,
Sonnhammer, E. L. L., and Bateman, A. (2008). The
Pfam protein families database. Nucl. Acids Res.,
36:D281–D288.
Gloor, G., Martin, L., Wahl, L., and Dunn, S. (2005). Mu-
tual information in protein multiple sequence align-
ments reveals two classes of coevolving positions.
Biochemistry, 44(19):7156–7165.
Hamacher, K. (2007). Information theoretical measures to
analyze trajectories in rational molecular design. J.
Comp. Chem., 28(16):2576–2580.
BIOINFORMATICS 2010 - International Conference on Bioinformatics
46

Hamacher, K. (2008). Relating sequence evolution of
HIV1-protease to its underlying molecular mechanics.
Gene, 422:30–36.
Hamacher, K. and McCammon, J. A. (2006). Computing
the amino acid specificity of fluctuations in biomolec-
ular systems. J. Chem. Theory Comput., 2(3):873–
878.
Higgins, D. G. and Sharp, P. M. (1988). Clustal: a pack-
age for performing multiple sequence alignment on a
microcomputer. Gene, 73(1):237–244.
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD
– Visual Molecular Dynamics. Journal of Molecular
Graphics, 14:33–38.
Lund, O., Nielsen, M., Lundegaard, C., and Brunak, C.
K. S. (2005). Immunological Bioinformatics. MIT
Press, Cambridge.
Pan, C., Kim, J., Chen, L., Wang, Q., and Lee, C. (2007).
The hiv positive selection mutation database. Nuc.
Acids Res., 35:D371–D375(1).
Perelson, A. S., Neumann, A. U., Markowitz, M., Leonard,
J., and Ho, D. (1996). HIV-1 dynamics in vivo: virion
clearance rate, infected cell life-span, and viral gener-
ation time. Science, 271:1582–1586.
Perryman, A. L., Lin, J.-H., and McCammon, J. A. (2006).
Restrained molecular dynamics simulations of hiv-1
protease: The first step in validating a new target for
drug design. Biopolymers, 82(3):272–284.
Prajapati, D. G., Ramajayam, R., Yadav, M. R., and Girid-
har, R. (2009). The search for potent, small molecule
nnrtis: A review. Bioorganic & Medicinal Chemistry,
17(16):5744–5762.
Reiling, K., Endres, N., Dauber, D., Craik, C., and Stroud,
R. (2002). Anisotropic dynamics of the JE-2147-
HIV protease complex: Drug resistance and thermo-
dynamic binding mode examined in a 1.09 a structure.
Biochemistry, 41:4582.
Richman, D., Margolis, D., Delaney, M., Greene, W. C.,
Hazuda, D., and Pomerantz, R. J. (2009). The chal-
lenge of finding a cure for HIV infection. Science,
323:1304–1307.
Rong, L., Gilchrist, M. A., Feng, Z., and Perelson, A. S.
(2007). Modeling within-host HIV-1 dynamics and
the evolution of drug resistance: Trade-offs between
viral enzyme function and drug susceptibility. J. Theo.
Biol., 247:804–818.
Sarafianos, S. G., Das, K., Hughes, S. H., and Arnold,
E. (2004). Taking aim at a moving target: design-
ing drugs to inhibit drug-resistant hiv-1 reverse tran-
scriptases. Current Opinion in Structural Biology,
14(6):716–30.
Schreck, T., Bremm, S., Held, S., and Hamacher, K. (2009).
to be published.
Shannon, C. E. (1951). Prediction and entropy of printed
english. The Bell System Technical Journal, 30:50–
64.
Stone, J. (1998). An Efficient Library for Parallel Ray Trac-
ing and Animation. Master’s thesis, Computer Science
Department, University of Missouri-Rolla.
Thompson, J., Higgins, D., and Gibson, T. (1994).
CLUSTAL W: improving the sensitivity of progres-
sive multiple sequence alignment through sequence
weighting, position-specific gap penalties and weight
matrix choice. Nucleic Acids Res., 22:4673–4680.
Trylska, J., Tozzini, V., Chang, C., and McCammon, J. A.
(2007). HIV-1 protease substrate binding and product
release pathways explored with coarse-grained molec-
ular dynamics. Biophys. J., 92:4179–4187.
Tsygankov, A. Y. (2009). Current developments in anti-
HIV/AIDS gene therapy. Curr Opin Investig Drugs,
10(2):137–149.
W.H. Press et al (1995). Numerical Recipies in C. Cam-
bridge University Press, Cambridge.
Wlodawer, A. and Erickson, J. (1993). Structure-based
inhibitors of HIV-1 protease. Annu. Rev. Biochem.,
62(1):543–585.
Yoshimura, K., Kato, R., Yusa, K., Kavlick, M. F., Maroun,
V.and Nguyen, A., Mimoto, T., Ueno, T., Shintani, M.,
Falloon, J., Masur, H., Hayashi, H., Erickson, J., and
Mitsuya, H. (1999). JE-2147: A dipeptide protease
inhibitor (PI) that potently inhibits multi-PI-resistant
HIV-1. Proc. Natl. Acad. Sci., 96:8675–8680.
CO-EVOLUTION IN HIV ENZYMES
47
