
CONCEPTUAL MODELING OF HUMAN GENOME
MUTATIONS
A Dichotomy Between what we Have and What we Should Have
M. Ángeles Pastor, Verónica Burriel and Óscar Pastor
Research Center on Software Production Methods (ProS). DSIC. Universidad Politécnica de Valencia
Camino Vera s/n. 46007, Valencia, Spain
Keywords: Conceptual Mmodelling, Bioinformatics and information systems, Human genome modeling, Linking
genotype and phenotype.
Abstract: It is well-known in the bioinformatics domain that the millions of mutations and polymorphisms that occur
in human populations are potential predictors of disease and any other type of human health related
problems. Finding sound strategies for going from the Genotype to the Phenotype is probably the main
challenge of the modern bioinformatics. Only with the sound knowledge provided by the IS theory, a
systematic approach to large-scale analysis of Genotype-Phenotype correlations can be developed. The
conceptual expressiveness of a well-known and widely-accepted database that stores the current information
about genome mutations, Human Gene Mutation Database, is compared with the information that is relevant
from a purely conceptual modelling perspective, and the result from this comparison is reported.
1 INTRODUCTION
Nowadays, it is well-known in the bioinformatics
domain that the millions of mutations and
polymorphisms that occur in human populations are
potential predictors of disease and other type of
human health related problems. To understand the
relationships that exist between a genome structure
and its external representation is a main challenge in
that domain. Finding sound strategies for going from
the Genotype to the Phenotype is probably the main
challenge of the modern genetics and of its main aid
in these days, bioinformatics. It is our belief that this
problem could only be solved with a strong
Information Systems (IS) background.
In fact, any solution to this problem has to be
faced working intensively with the current sources
of genomic information. From an IS perspective, we
claim that a more precise IS study is strictly required
in order to identify relevant concepts, and to
adequately represent and exploit them in a whole
Conceptual Schema of the Human Genome (CSHG),
which can facilitate the task of going from the
genotype level to its associated phenotype.
Such a CSHG is being developed, where all the
different views -genome structure, mutation,
transcription and translation processes - are included
(Virrueta, 2009). After having projected the previous
ideas onto the mutation level –which is the focus of
this paper-, some relevant discrepancies have been
found between what we find in the existing
databases and what we believe that should be the
right conceptual representation of the relevant
information (Pastor, 2008). We face this problem in
the paper. After having modeled the CSHG, we
focus on its mutation view. Taking as reference a
well-known and widely-accepted database that
stores the current information about different types
of genome mutations, Human Gene Mutation
Database (HGMD) (Stenson, 2009), we capture the
underlying model used by this database.
That done, we compare the conceptual
expressiveness found, with the information that we
consider relevant from a purely conceptual modeling
perspective. Basically, we fix consistency problems,
and we indicate how our CSHG could take the
appropriate contents from HGMD, preserving its
main properties of providing a coherent set of sound
information, that could be properly exploited to
connect genotype with phenotype. The practical aim
of our work is to develop a prototype of a tool to aid
genetic scientists in one of the tasks performed in a
genetic medecine laboratory. The task is the
160
Ángeles Pastor M., Burriel Coll V. and Pastor Ó. (2010).
CONCEPTUAL MODELING OF HUMAN GENOME MUTATIONS - A Dichotomy Between what we Have and What we Should Have.
In Proceedings of the First International Conference on Bioinformatics, pages 160-166
DOI: 10.5220/0002741201600166
Copyright
c
SciTePress
-id_symbol <<oid>> : string
-id_HUGO : int
-official_name : string
-summary : string
-chromosome : short
-locus : string
Gene
-ord_num <<oid>> : short
-start_position : long
-end_position : long
-strand : string
Allele
-Variant
*
-{id} 1
-sequence (derived) : string
Allelic Variant
-sequence : string
Allelic Reference Type
-id_allele_db : string
Allele Data Bank Identification
*
-{id}
1
-id_gene_db : string
Gene Data Bank Identificacion
1..*
-{id}
1
-id <<oid>> : string
-name : string
-description : string
Data Bank
1..*
-{id}
1
1..*
-{id}
1
-id_variation : int
-description : string
-id_variation_db : string
-type : string
-location : string
Variation
-Changes
*
0..*
-Obtained
1
*
1
*
-Referred
*
1
-description : string
Imprecise
Chromosomal
-type : string
Mutant
-identificacion<<oid>>
-description : string
Chrom. Mutation
1
-Influences
1..*
Location
Description
-id <<oid>>
-title
-authors
-abstract
-publication
Bibliography Ref
-bibliography Name DB <<oid>>
-URL
Bibliography DB
*
-{id}
1
-sequence : string
-origin : string
-position : int
Regulatory
Effect
-sequence : string
-repetition : int
Insertion
-bases : int
Deletion
-ins_sequence : string
-ins_repetition : int
-del_bases : int
Indel
-bases : int
Inversion
-original_data : string
-cause : string
-correction_date
Problematic load
Load
-id_syndrome : int
-name : string
Phenotype
-level : string
Certainty
1..*
-{id}
1
1..*
-{id}
1
-position : int
Precise
*
*
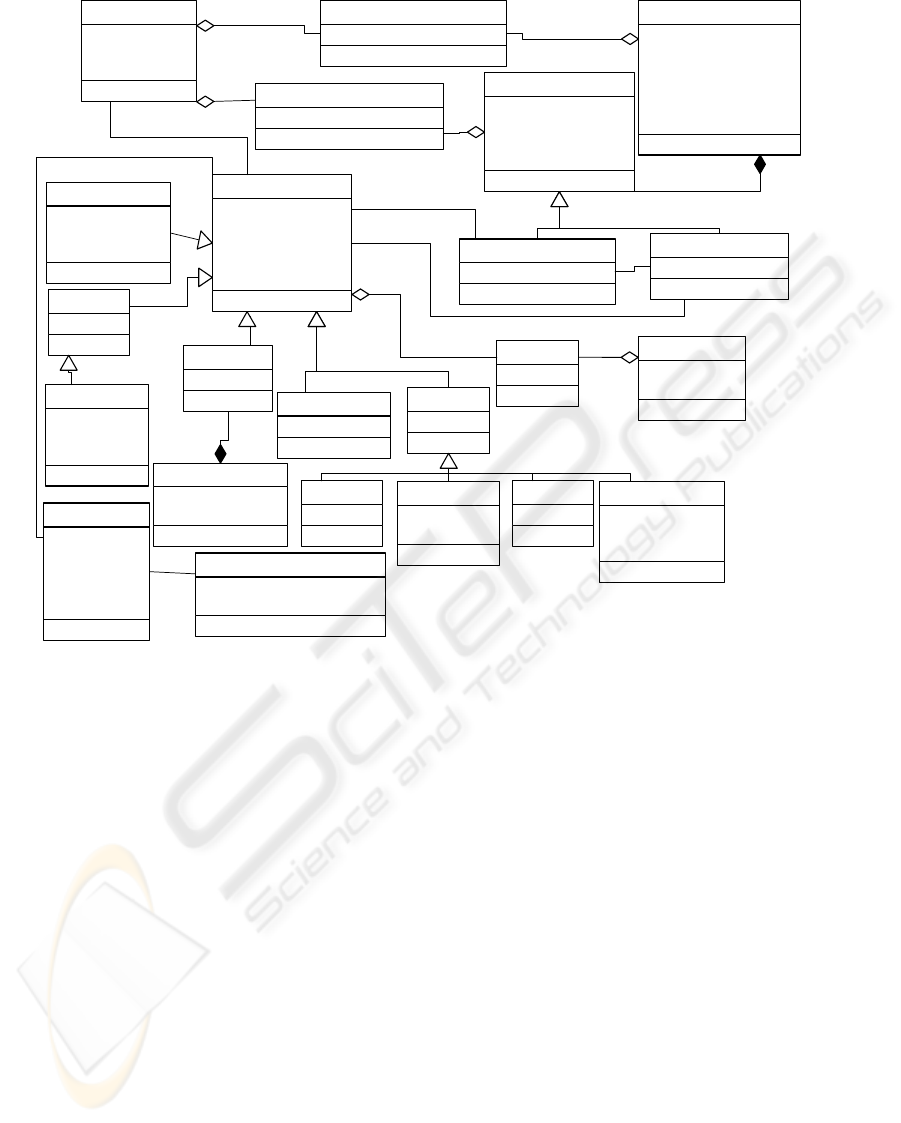
Figure 1: Mutation view of the CSHG.
following: given a sequence of a gene from a human
sample, obtain as a result the sequence variation(s)
present, if any, in the sample sequence with respect
to a given reference, as well as the links to the
relevant literature references about such variation(s).
In section 2 we describe the Mutation view of our
CSHG. In section 3 we explore the HGMD
structure. Section 4 performs the critical comparison
between the two models.
2 CSHG: MUTATION VIEW
Our proposal of CSHG has evolved from its
beginning through different versions. A version,
previous to the actual one, has been exhaustively
described in (Virrueta, 2009). In this paper, a new
version is presented, and its new features explained
with respect to the previous version. The main
differences are located in the Gene-Mutation view,
and it is in this one where this work focuses, because
the data from HGMD are specially related -although
not exclusively, to this view.
In the Figure 1 the knowledge about genes, their
structure and their allelic variants is modeled. The
principal classes in this view are Gene class, Allele
class and Variation class.
The Gene class models the concept of generic
gene independently of individual samples registered
in the databases. In this class, the relevant attributes
are the following: Id_HUGO, a code (HUGO) which
represents the universal code for the gene according
to the Human Genome Nomenclature Committee
(HGNC); this attribute is also the gene identification
in our conceptual model; Id_symbol, a symbol for
the gene according to HUGO; official_name, the
name of the gene; summary describes gene function;
chromosome, is the chromosome number where the
gene is located, and locus represents the gene
location into the chromosome.
The Allele class represents the different forms
that a gene can present in the nature. This class
contains the following attributes: ord_num attribute,
the internal identification number of the allele;
CONCEPTUAL MODELING OF HUMAN GENOME MUTATIONS - A Dichotomy Between what we Have and What
we Should Have
161

start_position and end_position respectively
describe the beginning and end of the allele respect
to the chromosome, and strand stands for one of the
two DNA chains (plus or minus).
The relationship between Gene class and Allele
class helps to identify an allele of a gene in the
information system. It allows that a gene may not to
have any allelic information.
The DataBank class represents different public
databases used to load our database. The
GeneDataBankIdentification class is the gene
identification in different public databases. The
AlleleDataBankIdentification class is the allele
identification in different public databases.
The Allelic Variant class and the Allelic
Reference Type class are specialized classes from
Allele class. The Allelic Reference Type class
represents the alleles which are used as reference in
the consulted data sources, while the Allelic Variant
represents allelic variations of a reference allele.
Both of these classes include a sequence attribute,
but while for the Allelic Reference Type class this
attribute stores the complete DNA sequence of the
allele, for the other it can be obtained by derivation
in a way that is explained later. Related is an
association between Allelic Reference Type and
Allelic Variant representing the existing relation
between a reference allele and its variations.
The main innovation in this CSHG version is the
Variation class. It represents the changes shown by
different DNA sequences compared with a reference
type. This class consists of the following attributes:
id_variation, a local identifier, description, a
description of the variation, id_variation_db, the
identifier given to the variation by the database
where it has been taken from, type which ranges
over the values‘mutant’, ‘neutral polymorphism’ and
‘unknown consequence’, and location which can be
‘chromosomal’ or ‘genic’.
The Variation class is associated with Data Bank
class and with Bibliography Reference; it is also
associated with the Allelic Reference Type class
through the Referred association as well as with the
Allelic Variant class by the Changes association; this
one represents that each variation is necessarily
shown by an allelic variant with respect to an allelic
reference type. The Changes association allows for
an allelic variant to include several variations with
respect to an allelic reference type; subsequently,
now it can be seen clearly how the sequence of the
Allelic Variant class can be derived if all the
variations respect to the Allelic Reference Type are
known; conversely, the variations of an Allelic
Variant respect to its Allelic Reference Type can be
derived if the two allelic sequences are known. As a
result of the new class, the specialization hierarchy
describing different types of valid variations belongs
to the Variation class, instead of to the Allelic
Variation class as it was in the previous version.
This specialization hierarchy classifies variations by
different criteria represented in four specializations:
• Location specialization represents
whether the variation affects only one
gene or a part of the chromosome.
• Description specialization models the
degree of knowledge of the variation.
• Effect specialization depends on the effect
on the phenotype.
• Load specialization for the variations
where data inconsistencies have been
found at the source.
The Chromosomal specialized class includes the
variations which affect to more than one gene. The
Chrom.Mutation class describes the chromosomal
variation. In the Description specialization, the
variation is classified in Imprecise and Precise.
When details about the variation are not known, it is
classified as imprecise. There is a description
attribute in the Imprecise class. When a variation is
precise its position is represented in the position
attribute, and the variation is classified in one of the
four specialized classes. If the precise variation type
is Insertion, a sequence (sequence attribute) has
been introduced n times (repetition attribute). If the
type is Deletion, certain nucleotides were deleted
from the specified position. When the type is Indel,
there was a deletion of
n nucleotides and then an
insertion of m nucleotides p times occurred.
Inversion means that the nucleotide chain flipped at
the position specified in the position attribute. In the
Effect specialization, variations with mutant effect
on the phenotype are represented in the Mutant
specialized class. They have a type attribute ranging
on values ‘splicing’, ’missense’, ’regulatory’ and
’others’. The regulatory variations have three
attributes: sequence which contains the substitution
sequences; origin denotes the point from which the
relative position of the mutation is given. Finally, by
the Load specialization, variations are classified in
Problematic Load when the data from sources have
inconsistencies. It has three attributes: original_data,
where the incorrect data is stored; cause, an
explanation about the inconsistence found and
correction_date, empty while the inconsistence is
not repaired, and containing a date value indicating
when the inconsistence has been repaired.
The Phenotype class represents the different
external features that can be associated to variations;
BIOINFORMATICS 2010 - International Conference on Bioinformatics
162
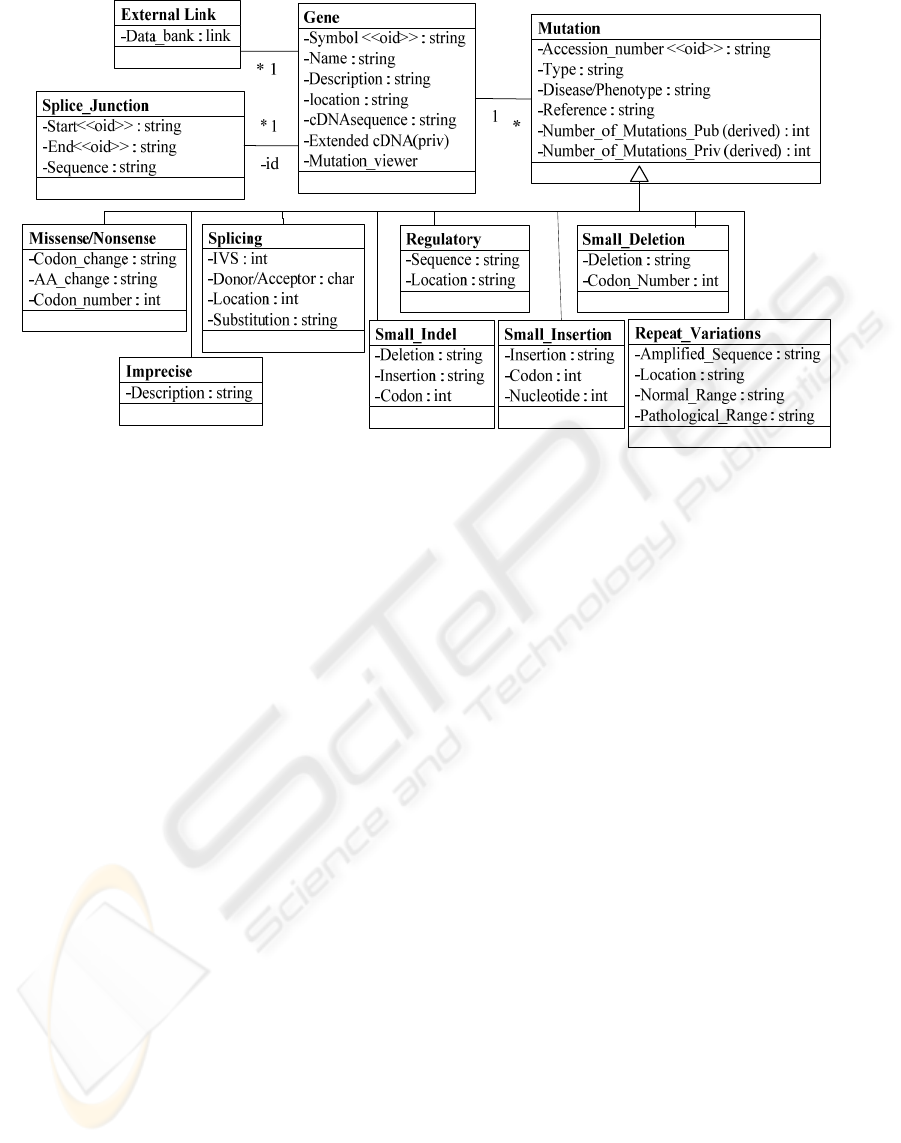
Figure 2: Conceptual schema deduced for HGMD®.
it has two attributes: id_syndrome, a code for the
phenotype and name, with the name or description
of the phenotype. The level of certainty about
whether a relationship exists between instances from
this two classes is represented by Certainty class
with the attribute level.
The rest of the Mutation view of the CSHG is not
affected by the comparison realized in section 4.
3 HGMD
®
: CONCEPTUAL VIEW
The HGMD
®
is a comprehensive core collection of
germ line mutations in nuclear genes that underlie or
are associated with human inherited disease
(Stenson, 2009). The information is accessible via
internet at different levels, one of them being public,
which is the subject of this study. It is presented in a
gene-wise basis and access to the subcategorized
mutation data is available from each gene page.
The complete information that can be publicly
found in HGMD about genes has been explored,
with the aim of inferring its conceptual
schemawhich is depicted in Figure 2.
The main class in the schema is Gene. A gene is
identified by its symbol attribute; its name is
included in the name attribute, the description
attribute includes a summary of the gene function,
the location attribute contains the gene position in
the chromosome, and a link to obtain the reference
cDNA (the cDNA attribute) is also found. Two
derived attributes number_of_mutations_Pub and
number_of_mutations_Priv show the total number of
public and private mutations respectively.
The splice_junction class each of the splice
junctions given for some genes. this information is
presented as a unique text. Each splice junction can
be extracted from it and defined by the start and the
end attributes, which, together with the gene symbol,
identify it; a sequence attribute can also be found;
with over 25 base pairs of exonic sequence, along
with 25 base pairs of intronic sequence.
The external_link class are the links associated to
each gene that provide information about it in
various databases like Genome DataBase (GDB) and
Online Mendelian Inheritance in Man (OMIM)
(Hamosh, 2005), among others.
Mutation class represents the generic information
of each mutation. They are identified by an
accession_number, and are also described by the
attributes type, disease/phenotype and reference_
text. The type attribute domain consists in ten
different values: ‘missense/ nonsense’, ‘splicing’,
‘regulatory’, ’small_ deletion’, ‘small_insertion’,
‘small_indel’, ‘gross
_ deletion’, ‘gross_insertion’,
‘complex_rearrangement’ and ‘repeat_variation’.
The disease/phenotype attribute contains the disease
or the phenotype that is associated to the mutation.
The reference_text attribute is the literature refe-
rence where the mutation has first been described.
Mutation class is specialized based on the type
attribute value. An exception is presented by the
Gross_Deletion, Gross_Insertion and Complex_
CONCEPTUAL MODELING OF HUMAN GENOME MUTATIONS - A Dichotomy Between what we Have and What
we Should Have
163

rearrangement values which are represented
altogether in the Imprecise class because they share
the same kind of information. Missense/Nonsense
class represents the mutation in which a single
nucleotide is changed and a non functional protein is
produced. Their attributes are: the codon_change
attribute, the aminoacid_change attribute, and the
codon_number attribute, that locates the affected
codon. The Splicing class describes mutations
affecting the mRNA splicing process. They include
the relative position of the lesion (location attribute)
with respect to a numbered intron (IVS attribute) at
the donor or acceptor splice site (donor/acceptor
attribute). The substitution attribute contains the
corresponding single base substitution. The
Regulatory class represents substitutions causing
regulatory abnormalities. The substitutions are
logged in its sequence attribute. The location
attribute indicates the position of the mutation
relative to the transcriptional initiation site, initiator
ATG or polyadenylation site. The Small_Deletion
class stands for deletions of 20 base pairs or less; the
deletion attribute contains the deleted bases. The
codon_number attribute shows the last complete
codon before the deletion. The Small_Insertion class
describes insertions of 20 base pairs or less. The
insertion attribute contains the inserted bases. The
number of the codon where the insertion locates is
found in the codon attribute, and the nucleotide
attribute describes the position of the first insertion
nucleotide. The Small_Indel class represents
insertions combined with deletions of 20 base pairs
or less. The deletion attribute contains the deleted
bases. The insertion attribute contains the inserted,
and the number of the codon where the insertions
locates is found in the codon attribute. The
Imprecise specialized class includes only the
description attribute which contains information
about the nature and location of each lesion. The
Repeat_Variation class includes some more
attributes such that amplified_sequence, containing
the repeated sequence, location, and the
normal_range and pathological_range attributes.
4 CSHG VERSUS HGMD
®
In this section. a critical comparison between the
two conceptual models CSHG and the one inferred
for the HGMD, is done, in order to analyze the
existing coincidences and differences between the
concepts included in each schema.
In the Gene class, which appears in both schemas,
most of the attributes are common in meaning. The
main significant difference is found for the DNA
sequence: while CSHG includes the complete
sequence for a gene, HGMD, only contains the
coding DNA. Small parts of the rest of DNA
sequence are also included in HGMD in the
sequence of the Splice_Junction class. This one is a
crucial difference, as HGMD lacks some DNA
segments which can be implied in mutations. CSHG
also includes in the Gene class the id_HUGO
attribute, not present in HGMD, and the
chromosome attribute is neither found in HGMD,
although it forms the beginning part of the locus
attribute..
The HGMD Splice_Junction class is not included
in CSHG as it is unnecessary, because it is
comprised in the complete genomic sequences of
CSGH Allelic Variant and Allelic Reference Type.
The comparison of the rest of the schemas is
some more complex. At a first glance it could seem
that the HGMD Mutation class has to be compared
with the CSHG Variation class, as they stand for a
similar information describing changes between
DNA sequences. But there is an important difference
between the two cited classes: Variation class
models any variation in an allelic DNA sequence
with respect to an allelic reference DNA sequence,
while HGMD Mutation class only refers to
pathological variations, that is, those variations
which produce or are associated with diseases. As a
consequence of this difference in the scope of the
variations, the HGMD Mutation class has also to be
compared with the CSHG Mutant specialized class,
specially in the aspects relative to their respective
specializations.
Both Variation and Mutation class have an
identifier, with different values because it is a local
identifier. In the HGMD Mutation class, the attribute
disease/phenotype indicates the disease or associated
to disease polymorphism produced by each mutation
which corresponds to the name attribute of the
Phenotype class in CSHG schema. When the
HGMD disease/phenotype attribute includes a
symbol representing the uncertainty of the data, in,
the CSHG Certainty class the level attribute is set to
‘doubtful’; otherwise, it is set to ‘sure’
Another difference is found in the coverage of the
bibliographic reference. HGMD includes an attribute
for the reference that originally described each
mutation, while the CSHG Variation class relates
with Bibliography Reference class through an
association that allows for several references for
each variation. Nevertheless, HGMD also includes
the External Link class where other databases can be
consulted to look for information about each gene.
BIOINFORMATICS 2010 - International Conference on Bioinformatics
164

In the HGMD Mutation class, the values of the
type attribute define the specialized classes in the
specialization of Mutation. In CSHG Mutation class
also an attribute for this information is found.
As described in section 3, the CSHG Variation
class is specialized attending to four criteria at a first
level, and two of the specialized classes, Mutant and
Precise, are affected by respective specializations at
a second level. On the other hand also the HGMD
Mutation class is specialized in several classes. As a
result of the comparison it is seen that the set of
specialized classes existing at the second level in
CSHG almost exactly match the set of specialized
classes of the HGMD Mutation class. Only a change
is found consisting in the CSHG Inversion class
instead of HGMD Repeat Variations. In CSHG, the
repeat variations are treated together with insertions
in the Insertion class, where the ins_repetition
attribute allows for a 1 value when it represents a
common insertion or a greater than 1 value if it is
used for a repeat variation.
From this point forward, the representation of
mutant variations diverge, as HGMD locates the
mutations in the coding DNA, or referred to the
splice junctions or to some special sites in the
chromosome, due to the absence of absolute
coordinates. Conversely, CSHG uses absolute
chromosome positions. For this reason, the attributes
do not coincide, specially for those specializations
not allocated in the coding DNA. CSGH Precise
class describes all the precise variations as
insertions, deletions, indels or inversions, whose
attributes allow for a complete description. By other
hand, HGMD separates from this description the
regulatory and the splicing mutations although they
could have been considered as indels. It has to be
done in this way because these variations occur out
of the coding DNA, so they have to be described in a
different way. The missense/nonsense mutations are
also represented in a different specialization, as they
informe about the amino acid change produced by
the mutation.
5 RELATED WORK
The presence of a sound conceptual modeling
background is not too frequent in the context of
Bioinformatics when we talk about sound
conceptual schemas of information related to the
genome and how to load their corresponding
database with the appropriate contents. While
obtaining the full DNA sequence of a diploid
genome of a single individual is becoming more and
more feasible (Wheeler, 2008), its semantic
interpretation has to be considered unachievable by
now. To compare such a full DNA sequence to a
given reference genome is currently an extremely
vast task. Only in (Wheeler, 2008), the performed
comparison led to the identification of 3,3 million
single nucleotide polymorphisms (SNPs), of which
up to 10,654 cause amino-acid substitution within
the analyzed coding sequence. To understand
precisely the relation between these genotype
aspects and their phenotype projection based on
conceptual models as the key artifact is the main
goal of our work. Until what we currently know, this
constitutes an original perspective in that field.
Other relevant works focus on the large number
of uncharacterized SNPs and potential mutations, to
drive the development of computational methods
aimed at identifying those variations likely to cause
disease (Yandell, 2008). A collection of known
disease-causing variations drawn from OMIM
(Hamosh, 2005) and HGMD (HGMD) are mapped
to their gene annotations and protein sequences, in
order to identify and characterize pairs of variations
that occur at homologous positions within human
disease genes. Again, it is our opinion that this work
is too solution-space centered, meaning by that that
it is developed around specific data coming from
particular data banks that –as we have shown in this
paper- don’t always offer a consistent view of the
involved data. Our claim is that these approaches
lack a core conceptual schema that should act as an
effective central repository for the semantically
relevant information.
Our work has been based on some pioneer work
(Paton, 2000). While it provides an initial sketch of
what a genome conceptual model should include, we
focus on the concrete view of mutations to develop
in detail these ideas, comparing from a modeling
perspective what we should expect to find according
to the conceptual schema with what the existing data
sources really provide.
Our aim is to set a sound basis to facilitate the
progress of genomics understanding and its effective
use in this conceptual-modeling-based direction.
6 CONCLUSIONS
When the problem of modeling the Human Genome
is faced from a pure top-down conceptual model
perspective, the relevant concepts are properly
represented in a conceptual schema. When this
schema has to be loaded with the adequate contents,
we have to go to the existing biological data sources,
as we have done with HGMD for the Mutation view
CONCEPTUAL MODELING OF HUMAN GENOME MUTATIONS - A Dichotomy Between what we Have and What
we Should Have
165

of the Genome. The problem analyzed in this paper
is that we do not find there what we supposed to
find, because there is a conceptual mismatch
between what we represent as relevant conceptual
primitives in the schema, and what the data sources
provide.
There is for sure a set of common information,
but we have discovered a very relevant set of
differences, that is basic to precisely identify to be
able to incorporate the right contents in the database
that corresponds to the CSHG
Summarizing, the information provided by
HGMD is very useful for the study of mutations.
The web presentation is very clear and easy to use,
and the amount of described mutations is high.
Nevertheless, the presented classification for
mutations seems to be guided by the lack of absolute
coordinates in a genomic reference. In this sense, the
hierarchy defined for the Variation class in CSHG is
much more complete, and obeys to conceptual
criteria. Also the way in which the mutation is
described in HGMD is a problem, using a character
string with uppercase and lowercase letters, and rare
symbols to properly define the mutation location.
Even so, in some cases the exact situation cannot be
found due to the same lack. Furthermore, due to the
deep analysis realized to the HGMD database, some
data inconsistencies have been found and reported to
the HGMD managers so they can amend them.
Another contribution of this paper is the process
that can be applied to incorporate correct contents
from other biological data sources selected
according to their data reliability. This would
provide a structured way to solve the very important
problem of data heterogeneity that the treatment of
genomic information suffers nowadays, mainly due
to the fact that too many data silos exist, with too
much independent and sometimes even inconsistent
information that is really hard to manage efficiently.
Having a database that has been precisely defined in
terms of a corresponding concrete conceptual
schema, and defining the adequate mappings
between selected parts of existing biological data
sources and their corresponding representation in the
CSHG database, a whole, consistent human genome
data repository would be available to be exploited.
This paper provides a concrete step in that direction.
Also, as a practical result of this work, a
prototype of a tool has been developed using the
database defined from the CSHG; to aid genetists in
the laboratory to fill their reports on gene variations
in a small fraction of the used time when the work is
done without this tool.
ACKNOWLEDGEMENTS
The authors thanks to the members of the PROS
Center Genoma group M.Pérez-Alonso, A. M.
Levin, J. C. Casamayor, M. Celma, L. Mota, F.
Valverde, A. M. Martínez, M. Van Der Kroon, I.
Lereu, A. Virrueta and L. E. Eraso for fruitful
discussions.
REFERENCES
George R. A., Smith T. D., Callaghan S., Hardman L.,
Pierides C., Horaitis O., Wouters M. A. and Cotton
R.G.H., 2007. General mutation databases: analysis
and review. In J. Med. Genet. published online 24
Sep; doi:10.1136/jmg.2007.052639, http://jmg.bmj.
com /cgi/rapidpdf/jmg.2007.052639v1.pdf.
Hamosh A. et als., 2005. Online Mendelian Inheritance in
Man (OMIM), a knowledge base of human genes and
genetic disorders. In Nucleic Acids Research 33:D514-
D517.
HUGO Gene Nomenclature Committee, http://www.gene
names.org.
Pastor, O. 2008. Conceptual Modeling Meets the Human
Genome. In Conceptual Modeling-ER 2008, Li Q.,
Spaccapietra S., Yu E. and Olivé A. (eds.). LNCS, vol
5231, pp 1-11 Springer, Berlin Heidelberg.
Paton W.N., Khan S., Hayes A., Moussouni F., Brass A.,
Eilbeck K., Globe C., Hubbard S., Oliver S., 2000:
Conceptual modeling of genomic information. In
Bioinformatics. 16, 6, 548–57.
Stenson et als. 2009, The Human Gene Mutation Database
(HGMD®): 2008 Update. In Genome Med (2009)
1(1):13
The Human Gene Mutation Database at the Institute of
Medical Genetics. Cardiff,, http://www.hgmd.cf.ac.uk
Virrueta A., et al. (2009): Enforcing Conceptual Modeling
to Improve the Understanding of Human Genome.
Poster and Demo Proceedings DILS 2009. pp. 29.
Wheeler D. A. et als., 2008. The Complete Genome Of An
Individual By Massivelly Parallel DNA Sequencing.
In Nature. Vol. 452/17, April 2008, 872-877.
Yandell M. et als., 2008. Genome-Wide Analysis of
Human Disease Alleles Reveals That Their Location
Are Correlated in Paralogous Proteins. In PLoS
Computational Biology, Vol.4(11):e1000218, doi:
10.1371/journal.pcbi.1000218.
BIOINFORMATICS 2010 - International Conference on Bioinformatics
166
