
IMPROVING SEARCH FOR LOW ENERGY PROTEIN
STRUCTURES WITH AN ITERATIVE NICHE GENETIC
ALGORITHM
Glennie Helles
University of Copenhagen, Department of Computer Science, Universitetsparken 1, 2100 Copenhagen, Denmark
Keywords:
Protein structure prediction, Parallelism, Genetic algorithm, Parallel tempering.
Abstract:
In attempts to predict the tertiary structure of proteins we use almost exclusively metaheuristics. However,
despite known differences in performance of metaheuristics for different problems, the effect of the choice
of metaheuristic has received precious little attention in this field. Particularly parallel implementations have
been demonstrated to generally outperform their sequential counterparts, but they are nevertheless used to a
much lesser extent for protein structure prediction. In this work we focus strictly on parallel algorithms for
protein structure prediction and propose a parallel algorithm, which adds an iterative layer to the traditional
niche genetic algorithm. We implement both the traditional niche genetic algorithm and the parallel tempering
algorithm in a fashion that allows us to compare the algorithms and look at how they differ in performance.
The results show that the iterative niche algorithm converges much faster at lower energy structures than both
the traditional niche genetic algorithm and the parallel tempering algorithm.
1 INTRODUCTION
Metaheuristics are known to perform well on high-
complexity problems where the search space becomes
too big for exhaustivesearch to be feasible. Prediction
of the three-dimensional structure of proteins from
their primary sequence alone, known as ab initio or
textitde novo folding
1
, is such a problem and meta-
heuristics are almost exclusively used to solve this
problem (Helles, 2008; Oakley et al., 2008).
Proteins are made up by amino acids that are
strung together like pearls on a string such that each
amino acid is connected to its neighboring amino
acid(s) via a peptide bond, ω. However, despite the
rigid nature of the peptide bond, atoms can in theory
rotate almost freely around the two other backbone
bonds – the N–C
α
bond, φ, and the C
α
–C’ bond, ψ
1
The term ab initio traditionally refers to prediction
methods that start without any knowledge of any globally
similar folds thereby setting them aside from homology
modeling techniques. However, many so called ab initio
methods do in fact use secondary structure prediction algo-
rithms that are trained from knowledge of already known
structures, or they use fragment assembly compiled from
known structures. Some choose to refer to this as de novo
prediction rather than ab initio prediction. The term ab ini-
tio will be used in this publication
– which means that just like a pearl necklace, a pro-
tein can be folded up in infinitely many ways, which
is the reason that protein structure prediction poses
such a big problem. Fortunately, steric clashes be-
tween atoms in neighboring amino acids do impose a
significant restraint on the flexibility of the φ and ψ
angles actually observed for amino acids (Ramachan-
dran and Sasisekharan, 1968), but searching exhaus-
tively for the structure with the lowest energy remains
elusive.
Judging from the literature, the Monte Carlo vari-
ant known as Simulated Annealing appear to be the
preferred meta-heuristic for ab initio structure predic-
tion followed by Genetic Algorithms (Helles, 2008).
Both metaheuristics can be parallelized, and the par-
allel versions are generally believed to perform bet-
ter in rugged energy landscapes like those associated
with protein structure prediction (Earlab and Deem,
2005). Oddly enough, the parallel versions are never-
theless used to a much lesser extent than their sequen-
tial counterparts in the field of protein structure pre-
diction. We speculate that that is mostly because the
effect of the choice of metaheuristics has been paid
little attention in this field that is notoriously haunted
by many other fundamental problems. The most sig-
nificant obstacle probably being finding an appropri-
ate energy function that can be used to score a protein,
226
Helles G. (2010).
IMPROVING SEARCH FOR LOW ENERGY PROTEIN STRUCTURES WITH AN ITERATIVE NICHE GENETIC ALGORITHM.
In Proceedings of the First International Conference on Bioinformatics, pages 226-232
DOI: 10.5220/0002743702260232
Copyright
c
SciTePress

which has by far received the most attention over the
years.
To our knowledge there exists only one parallel
version of the Simulated annealing algorithm known
as the Parallel Tempering or Monte Carlo Replica Ex-
change algorithm (Swendsen and Wang, 1986; Earlab
and Deem, 2005). In Parallel tempering (PT) many
simulations, or replicas, are started and run in paral-
lel. The solutions are sampled in the same fashion as
in the regular simulated annealing approach by mak-
ing small alterations to the solution and accepting the
change with a certain probability. However, instead
of lowering the temperature like in the simulated an-
nealing approach the simulations are run at different
but steady temperatures throughout the search. Two
replicas may be swapped with a probability that de-
pends on both differences in energy and temperatures,
such that a replica running at a lower temperature can
be exchanged with a replica running at a higher tem-
perature, thereby giving replicas a greater chance of
overcoming local minima barriers.
For genetic algorithms there exists several parallel
variants that generally offer significant improvements
by converging faster at often better solutions than the
non-parallel version. There are two major approaches
to parallelizing genetic algorithms. One is often re-
ferred to as the master-slave model, where a single
process (the master) controls the genetic algorithm,
but uses a number of other processes (the slaves) to
evaluate and possible breed the individuals. The slave
processes are run in parallel.
The other parallelization paradigm frequently
used is the niche model (also known as the island
hopping or deme model). A niche genetic algorithm
(nGA) is an implementation where several instances
of a genetic algorithm are run in parallel, evolving
sub-populations independently from each other (the
different niches). At certain points during evolution
individuals migrate to other niches and become part
of the population of that niche. The major advan-
tage of nGAs is that they not only allow evolvement
of multiple solutions at the same time, they exploit
the fact that different runs of the same genetic algo-
rithm is likely to produce different suboptimal solu-
tions, that combined are likely to yield better results.
Like PT the advantage of nGA is expected to be more
profound when the fitness landscape is very rugged.
PT and nGA, with N replicas and niches re-
spectively, essentially requires N times more com-
putational time than a single run of their sequential
counterparts. However, with multi-core computers
and CPU clusters being readily available to most re-
searchers, they can be executed in parallel and the ex-
tra computational time required does thus not impose
a problem. On top of that, PT and nGA generally
search more efficiently and usually arrive at much bet-
ter results, which makes the parallelization of these
meta-heuristics an attractive feature indeed.
In this paper we propose an iterative variant of a
nGA, called inGA (iterative niche genetic algorithm)
for protein structure prediction. The algorithm is de-
signed to increase search efficiency by locating and
converging on the low energy structures faster than
both nGA and PT. Essentially, the strategy corre-
sponds to letting all niches converge before migrating
individuals between them and restarting as described
by Cantu-Paz and Goldberg (Cant-Paz and Goldberg,
1996). However, while running each niche to conver-
gence worked well for the problem instances chosen
by Cantu-Paz and Goldberg, work by Heiler (Heiler,
1998) suggest that for protein structure prediction the
quality of predicted structures decrease when the in-
dividuals are locally optimized before the genetic op-
erators are applied. Rather than running to conver-
gence we thus suggest a kind of early stopping, which
generate low energy structures without spending too
much time on refining suboptimal structures.
2 METHODS
In the traditional niche genetic algorithm (nGA),
evolvement of several populations are run in parallel
and completely independent from each other. At cer-
tain points individuals from one or more niches (is-
lands or demes) are chosen according to some selec-
tion strategy and migrated to other niches, where they
replace individuals also chosen according to some se-
lection strategy. Usually the selection strategies are
based on the fitness values of the individuals such that
the best individuals from one niche are migrated to
another niche where they replace the worst individu-
als, as this migration strategy yields the fastest con-
vergence (Alba, 2005).
We propose an iterative niche genetic algorithm
(inGA) that performs a type of elitist refinement. Like
the traditional niche genetic algorithm multiple pop-
ulations are initially created and evolved in parallel,
but unlike the traditional niche algorithms, individu-
als do not migrate to other niches. Rather we stop
evolvement of all populations after a predefined num-
ber of generations, g, and choose the best solution
from each of the n niches. The individuals not se-
lected are destroyed while the selected individual are
put together in a new population, pop. pop is then
cloned n times and the cloned populations are placed
on the n niches where evolvement of new (and ini-
tially identical) populations is then carried out for g
IMPROVING SEARCH FOR LOW ENERGY PROTEIN STRUCTURES WITH AN ITERATIVE NICHE GENETIC
ALGORITHM
227

generations. The procedure of stopping, selecting,
cloning and restarting is repeated until the algorithm
converges. The pseudo code for the algorithm is given
in Algorithm 1
Algorithm 1. Pseudo code for inGA.
niches ← CREATE THREADS(n)
pop ← NULL
while !DONE() do
for each n in niches do
if pop equals NULL then
population ← CREATE POPULATION()
else
population ← CLONE(pop)
end if
niches[n] ← start(GA(population, g))
end for
WAIT FOR COMPLETION(niches)
pop ← NULL
for each n in niches do
pop ← GET BEST INDIVIDUAL(niches[n])
end for
end while
This strategy requires the number of individuals
in each niche to be the same as the number of niches,
although a different strategy could, of course, also be
utilized. In this work we ran 20 parallel niches with
20 individuals in each niche.
We did preliminary experiments to determine how
many generations to run the niches in each iteration.
We wanted the GA to run just long enough to reach
a good solution that captured the best traits of that
niche and by analyzing the development of the en-
ergy we found that by far the largest improvements
happen during the first 100 generations. We thus set-
tled at running 100 generations per iteration. We did
try to run the GA for 200 generations, but found that
while the initial niche solutions were improved,the fi-
nal result was not, which is much in keeping with the
findings presented in (Heiler, 1998).
The selection strategy employed both an elitism
strategy and the fitness proportionate selection strat-
egy known as a roulette wheel. The elitism strategy
clones and transfers the 10% top scoring individuals
unaltered to the next generation thereby ensuring that
the best individuals are always kept. However, the
10% best individuals are also allowed to compete in
the roulette wheel selection, where each individual is
chosen with a probability corresponding to its fitness
value. This strategy is chosen over rank selection to
ensure a better chance for low scoring individuals to
be selected.
Individuals selected by the roulette wheel are sub-
jected to crossover and mutation. A multi-point
crossover strategy is used where the number of
crossover sites, c, are chosen according to a Gaussian
distribution. The c actual crossover sites are chosen at
random. The advantage of multi-point crossover over
single point crossover is that it eliminates the bias of
the end segments that is commonly raised as an issue
with the vector representation employed by most ge-
netic algorithms. Also multi-point crossover typically
results in bigger alterations of the solutions causing
the genetic algorithm to explore very different regions
of the search space.
As is often the choice, the mutation rate is set
fairly low to a value of 0.001. Mutation is thus not
the driving force in the folding process, but is used
mainly as a way to introduce new genes into the ex-
isting gene pool.
It should be noted that setting the hyper-
parameters of genetic algorithms (such as selection
and recombination strategies) is an optimization prob-
lem in itself. We have looked to the literature for in-
spiration and carried out numerous experiments with
different combinations before settling on the ones de-
scribed here.
2.1 Encoding
In this work we use a physics based energy poten-
tials, called POISE (Lin et al., 2007), which requires
a full atom model. However, rather than searching
the Cartesian space, only dihedral angles, bond angles
and bond lengths (referred to as the set of structural
variables) are explicitly represented as atom positions
can be calculated directly from these using standard
matrix operations. A protein is thus encoded as a vec-
tor (chromosome) of S
structvar
, where each S
structvar
represents the structural variable of one amino acid.
One of the problems often encountered during
encoding of proteins is the occurrence of clashing
atoms. When two atoms come within very close prox-
imity, the laws of physic dictate that the energy will
rapidly grow towards infinity and the atoms will be
forced apart. When using a full atom model one of
two strategies can be utilized; either one can explic-
itly check and make sure that atoms do not clash or
clash are tolerated, but heavily penalized by the en-
ergy function, such that solutions with clashing atoms
stand little chance of being accepted/selected. The lat-
ter works best in conjunction with statistics based en-
ergy function where the ’energy’ term is usually an
artificial pseudo energy made up from many param-
eters that are non-numerical in nature. With a pure
physics based energy potential, the infinitely large in-
crease in energy caused by two clashing atoms will
BIOINFORMATICS 2010 - International Conference on Bioinformatics
228

cause overflow on a computer. As we utilize a pure
physics based potential, a check for clashing atoms
is thus carried out, before a solution is evaluated and
only clash-free structures are accepted.
The protein is encoded sequentially and for ev-
ery new residue added we check whether the S
structvar
chosen causes any atoms of the new amino acid to
clash with atoms in the residues that have already
been added. If a clash occur a new S
structvar
for
the amino acid is chosen. If the problem with atom
clash has not been resolved after 20 different S
structvar
have been tried for the amino acid, we backtrack and
choose a new S
structvar
for the previous residue. Al-
though the theoretical running time for this strategy is
O(2
n
) the running time was not found to be an issue
in practice.
2.2 Move Set
The move set is defined as the set of possible com-
binations of bond lengths, angles and dihedral angles
for each amino acid. Theoretically, the move set is un-
restricted, but in practice we know that bond lengths
and angles vary very little and dihedral angles are
heavily biased towards certain areas of the dihedral
angle space. Here, we thus choose bond angles and
bond lengths for amino acids randomly from within a
small interval (up to ±0.1) of the optimal angles and
lengths as defined in the AMBER 99 parameters.
The dihedral angles space is likewise restricted.
In Fonseca and Helles (Fonseca and Helles, 2009) a
probability distribution is predicted for each amino
acid in a sequence by considering the neighboring
amino acids. This probability distribution is used here
such that the dihedral angles are chosen from this
sequence-dependent distribution thereby maximizing
the probability of sampling a realistic area of the di-
hedral angle space.
2.3 Energy Function
Generally speaking, energy functions can be divided
into two categories: physics based energy function
and statistics based energy functions. The energy
function used here, called POISE, is a purely physics
based potential, described in more details in (Lin
et al., 2007). It combines the AMBER force field
(Swendsen and Wang, 1986):
E
protein
=
bonds
∑
i
K
b
(b
i
− b
0
)
2
+
angles
∑
i
K
θ
(θ
i
− θ
0
)
2
+
dihedrals
∑
i
k
x
[1+ cos(nφ − γ)]+
+
N
∑
i
N
∑
i< j
q
i
q
j
r
ij
+ 4ε
ij
"
σ
ij
r
ij
12
−
σ
ij
r
ij
6
#!
(1)
with a Generalized Born component:
V
GB
= −
1
2
1
ε
p
−
1
ε
w
∑
i
∑
i< j
q
i
q
j
f
GB
ij
(r
ij
)
(2)
f
GB
ij
(r
ij
) =
"
r
2
ij
+ R
i
R
j
exp
−r
ij
(
2)
4R
i
R
j
!#
1
2
and a hydrophobic mean force potential:
V
HMFP
=
N
c
∑
i∈SA
i
>A
c
tanh(SA
i
)
N
c
∑
j∈SA
j
>A
c
, j6=i
tanh(SA
j
)
x
3
∑
k=1
h
k
exp
−
r
ij
− c
k
w
k
2
!
(3)
We refer to (Lin et al., 2007) for an explanation of the
parameters.
The potential considers interatomic energies be-
tween all pairs of atoms and the time complexity of
calculating the potential is thus quadratic (O(n
2
)).
Fortunately, the vast majority of atoms in a protein
are simply too far apart to exert any power on each
other and by simply omitting calculations of these in-
teractions, the function is implemented such that the
potential is calculated in linear time.
It should be noted, that in this current work we
are primarily concerned with testing the performance
of different parallel meta-heuristics. We have chosen
the POISE energy function because it gives a realistic
impression of the very rugged fitness landscape that
the algorithms have to navigate around
2.4 Test Proteins
A standardized, diverse set of proteins that is guar-
anteed to provide an adequate representation of pro-
tein structures does unfortunately not exist. The test
set used here is constructed such that 2 proteins from
each of the categories α, β and αβ have been picked
from the PDB Select 25%
2
. Only small to medium
sized proteins have been included. The smallest pro-
tein includes 46 amino acids and the largest protein
includes 81 amino acids (Table 1).
2
http://bioinfo.tg.fh-giessen.de/pdbselect/
recent.pdb select25
IMPROVING SEARCH FOR LOW ENERGY PROTEIN STRUCTURES WITH AN ITERATIVE NICHE GENETIC
ALGORITHM
229
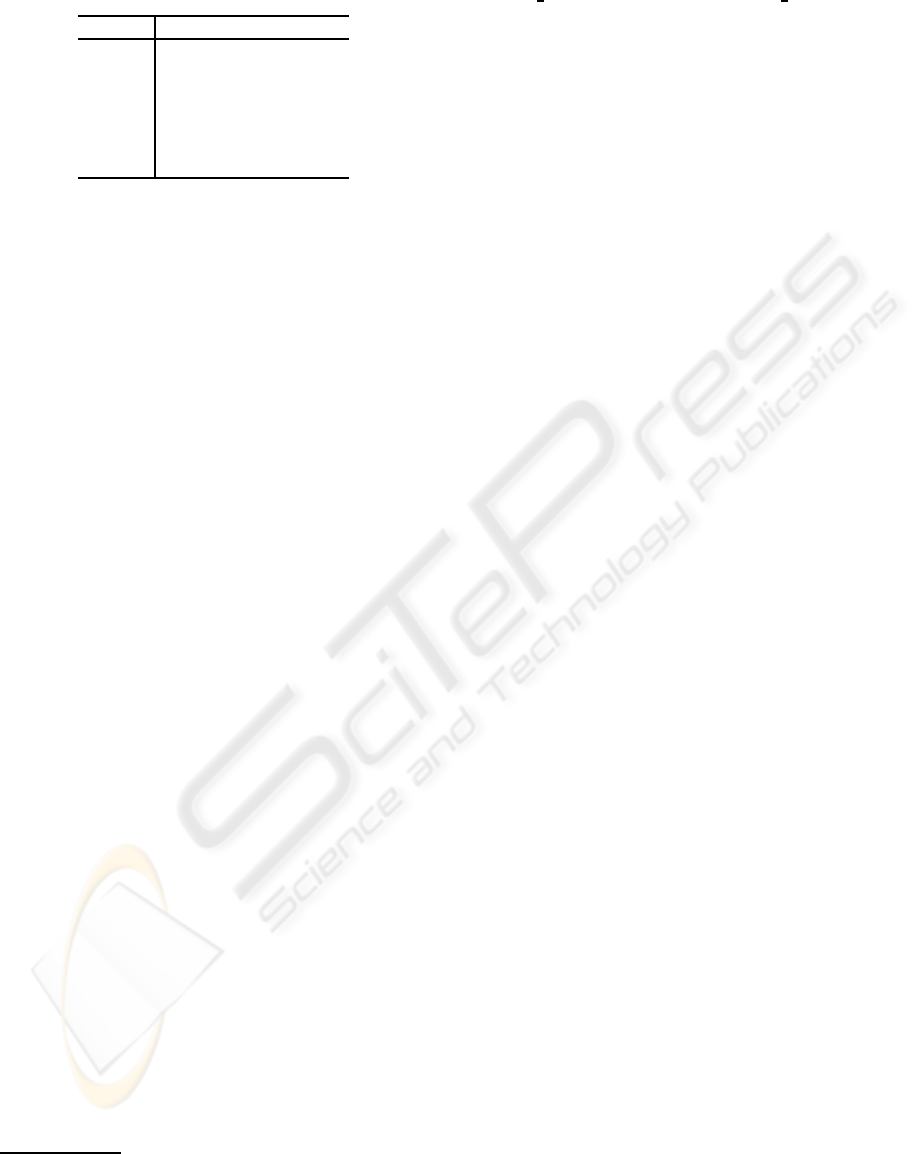
Table 1: Test proteins.
Category Residues
1C75 α 71
1NKD α 59
1YK4 β 52
2O9S β 67
1EJG αβ 46
1IQZ αβ 81
2.5 Benchmarking Algorithms
In order to evaluate the performance of inGA we
implemented both the traditional nGA and the par-
allel version of SA, called parallel tempering (also
known as the Replica Exchange Monte Carlo algo-
rithm). Parallel algorithms have numerous times been
reported to outperform the sequential algorithms, and
here we thus focus only on parallel algorithms
3
.
The nGA use the same GA to evolve populations
as in inGA, and like for inGA we also used 20 par-
allel niches in nGA. The convergence rate of a nGA
is strongly affected by the migration scheme (Alba,
2005). Migrating and replacing only a few randomly
chosen individuals leads to very slow convergence
whereas migrating the best and replacing the worst
leads to the fastest convergence. To make the com-
parison with inGA, which is highly elitist, fair, we
employed a rather strong selection scheme such that
every 100 generation we chose the 50% best individ-
uals from each population, cloned them and migrated
them to another niche where it replaced the 50% worst
individuals.
The PT algorithm is implemented such that it
utilizes the same encoding strategy, energy function
and move set as described above in order to ensure
comparability. The lowest and highest temperatures
were determined in the way proposed in (Sanvicente-
Snchez and Frausto-Sols, 2004), such that
c
highest
= −δE
max
/ln(P
A
(δE
max
)) (4)
and
c
lowest
= −δE
min
/ln(P
A
(δE
min
)) (5)
Initial experiments measuring differences in the
energy, E, between neighboring structures were run
to determine the values of E
max
and E
min
. P
A
(δE
max
)
and P
A
(δE
min
) were set to 0.95 and 0.05 respectively.
This resulted in c
highest
= 3800 and c
lowest
= 10 with
temperatures of the different replicas spaced accord-
ing to:
3
Although the results are not reported here, experiments
with both the standard non-parallel genetic algorithm and
simulated annealing was carried out as well, and they did
indeed perform worse than the parallel versions
temp replica
i+1
= 10∗ i ∗ 2+temp replica
i
(6)
The observed average probability of accepting a
swapping move between neighboring replicas was
roughly 20% in accordance with (Kone and Kofke,
2005), but with swapping of course occurring much
more frequently between replicas running at high
temperatures and much less frequently between repli-
cas running at low temperatures.
In each time step every replica goes through N
moves for a N-residue long protein. A move consists
of randomly selecting an amino acid and picking a
new S
structvar
for that amino acid. As is typical for the
simulated annealing approach a move from structure
s to some neighbor s
′
is accepted with the following
probabilities:
P(accept) =
exp
−(E
s
′
−E
s
)/K
b
T
E
s
′
≥ E
s
1 E
s
′
< E
s
(7)
where E is the energy of the structure and T is the
temperature.
We ran 20 parallel simulations. The probability
of accepting a swap between to replicas, i and j, was
given by:
P(i ↔ j) = min{1,exp
[−(β
i
−β
j
)(E
j
−E
i
)]
} (8)
where β is the inverse temperature β = 1/k
B
T and
β
i
> β
j
. Defining the probability of swapping repli-
cas such that it decreases exponentially as the gap be-
tween temperature increase is usually employed in PT
(Earlab and Deem, 2005) and also the reason why it
was chosen here.
3 RESULTS AND DISCUSSION
Early results from experimentation with the three dif-
ferent parallelization schemes on the test proteins,
shown in Table 2, look very promising with inGA
quickly and consistently locating structures of lower
energy than both nGA and PT. Please note that we
have not calculated RMSD of the final structures, be-
cause as such the search algorithms are all oblivi-
ous to the concept of a native structures. They seek
merely to minimize energy as specified by the POISE
potential and in this experiment we are only inter-
ested in determining how efficient the different al-
gorithms are in finding low energy structures in the
highly rugged energy landscape associated with pro-
tein structure prediction energy functions. A differ-
ent energy function would most likely lead to differ-
ent (either better or worse) quality of the final struc-
tures in terms of RMSD to the native structure, but the
differences in how well the algorithms perform with
BIOINFORMATICS 2010 - International Conference on Bioinformatics
230

Table 2: Early results of the three algorithms. Energies are
calculated with the POISE potential. Lower energies are
better.
nGA inGA PT
1C75 319 287 513
1NKD 113 -14 200
1YK4 216 142 275
2O9S 385 223 875
1EJG 4 -7 30
1IQZ 414 348 665
respect to each other would (expectedly) remain the
same.
Given unlimited time, all meta-heuristics would
probably find the same low energy structures. Un-
fortunately, time is usually not unlimited in practice
and designing search algorithms that increases search
efficiency such that we can obtain better results faster
becomes important. Parallelization has in itself in-
creased search efficiency, but from the results pre-
sented here it is evident that how the algorithms are
parallelized can also have a profound impact on how
efficiently the algorithms travel the energy surface in
their search for the global minima.
The solution space for a given protein sequence
is infinitely big and the key to success for a meta-
heuristics is usually a good balance between explo-
ration and exploitation. Minima should be explored
thoroughly while still allowing the algorithm to move
relatively freely across energy barriers. PT, nGA and
inGA all differ from each other in this exploration-
exploitation balance.
In PT the balance between exploration and ex-
ploitation is kept by running parallel simulations at
different temperatures. The advantage of PT is that
it can be run for exactly as long as time permits, be-
cause while it may settle at a minima, it does not re-
ally converge but rather keep exploring for a preset
number of iterations or until it is interrupted. As such
the PT algorithm enjoys the same theoretical guaran-
tee of finding the global minima as the simulated an-
nealing algorithm. However, while parallel tempering
reaches low energy structures faster than sequential
Monte Carlo simulations (Earlab and Deem, 2005),
the number of replicas used depend not so much on
available processors, but on what makes sense in or-
der to maintain proper communication between the
different replicas. In other words, there appears to
be an upper limit on what we can expect to gain in
performance that depend on the problem and not on
CPU power. For proteins of the length used here,
20 replicas ensure appropriate communication across
temperatures, and more replicas would thus only in-
crease the level of communication thereby setting off
the exploration-exploitationbalance, which would not
be desirable. Of course, for larger proteins where the
energy span between different structures is likely to
be greater than for the proteins used here, more repli-
cas would most likely be required to ensure proper
communication.
One of the reasons why PT does not reach the low
energy structures as fast as the genetic algorithms is
that although many replicas are run at the same time
they do not exchange information between replicas.
If a good solution is encountered at one temperature
it may be exploited by swapping it to a lower temper-
ature, but it does not share its favorable characteris-
tics with any of the other replicas. Parallel tempering
would most likely reach the same results as achieved
by inGA, but we postulate that because of the lack
of information sharing it can generally be assumed to
take much longer.
The genetic algorithms on the other hand have a
high degree of information sharing via their crossover
operator, which explains why the genetic algorithms
reach the lower energy structures much quicker. The
migration scheme we have used here for nGA is fairly
aggressive to ensure faster convergence that would be
comparable with inGA. From the results it is evident
that while nGA finds lower energy structures than PT
it does not reach structures with energies as low as
inGA. It should also be noted that despite the aggres-
sive migration strategy, nGA does not fully converge
within 20 iterations for any of the test proteins, al-
though signs of convergenceis beginning to show. We
did initially experiment with a less aggressive migra-
tion scheme (that migrated only the best individual),
but energies were significantly worse after the 20 it-
erations than with the chosen migration scheme and
it did of course not come near convergence within 20
iterations.
An issues with inGA is that it may simply con-
verge prematurely. The iterative strategy of inGA is
highly elitist and with 20 niches it usually converges
fully within 20 iterations for the small to medium
sized proteins used here. An elitist strategy (al-
ways picking the best) favors exploitation heavily and
will normally only work well in smooth energy land-
scapes. The energy landscape of proteins is obvi-
ously anything but smooth, but interestingly a bal-
ance with exploration does nevertheless appear to be
maintained in inGA by the niche approach. Explo-
ration can thus be controlled by simply adding more
or less niches. This is indeed a nice feature, since bet-
ter performance can then be brought to depend more
on available CPUs rather than on available time. Ob-
viously, adding more niches would most likely require
more iterations to fully converge,but the number of it-
IMPROVING SEARCH FOR LOW ENERGY PROTEIN STRUCTURES WITH AN ITERATIVE NICHE GENETIC
ALGORITHM
231

erations required to converge would expectedly grow
much slower for inGA than for nGA thereby mak-
ing the difference in performance between inGA and
nGA greater as the number of niches increase.
4 CONCLUSIONS
We presented an iterative variant of the parallel niche
genetic algorithm for protein structure prediction.
Early results show that the algorithm finds signifi-
cantly lower energy structures than both the tradi-
tional niche genetic algorithm and the parallel temper-
ing algorithm within comparable time. The algorithm
converges quickly and the exploration-exploitation
balance can be controlled with the number of niches
included, which means that search efficiency can be
expected to scale nicely with the number of available
CPUs.
REFERENCES
Alba, E. (2005). Parallel Metaheuristics. Wiley.
Cant-Paz, E. and Goldberg, D. E. (1996). Modeling ideal-
ized bounding cases of parallel genetic algorithms. In
In, pages 353–361. Morgan Kaufmann Publishers.
Earlab, D. J. and Deem, M. W. (2005). Parallel temper-
ing: Theory, applications, and new perspectives. Phys.
Chem. Chem. Phys., 7:3910.
Fonseca, R. and Helles, G. (2009). Predicting dihedral angle
probability distributions for protein coil residues from
primary sequence using neural networks. In submis-
sion with BMC Bioinformatics.
Heiler, M. (1998). Massively parallel gas for protein struc-
ture.
Helles, G. (2008). A comparative study of the reported per-
formance of Ab Initio protein structure prediction al-
gorithms. J. R. Soc. Interface, 5:387396.
Kone, A. and Kofke, D. A. (2005). Selection of temperature
intervals for parallel-tempering simulations. J. Chem.
Phys., 122:206101.
Lin, M. S., Fawzi, N. L., and Head-Gordon, T. (2007).
Hydrophobic potential of mean force as a solvation
function for protein structure prediction. Structure,
15:727–740.
Oakley, M. T., Barthel, D., Bykov, Y., Garibaldi, J. M.,
Burke, E. K., Krasnogor, N., and Hirst, J. D. (2008).
Search strategies in structural bioinformatics. Current
Protein and Peptide Science, 9:260274.
Ramachandran, G. N. and Sasisekharan, V. (1968). Con-
formations of polypeptides and proteins. Adv. Protein
Chem., 23:283–437.
Sanvicente-Snchez, H. and Frausto-Sols, J. (2004). A
method to establish the cooling scheme in simulated
annealing like algorithms. LNCS, 3945:755–763.
Swendsen, R. H. and Wang, J.-S. (1986). Replica monte
carlo simulation of spin-glasses. Physical review let-
ters, 57:2607–2609.
BIOINFORMATICS 2010 - International Conference on Bioinformatics
232
