
STRUCTURAL MOTIF ENUMERATION IN
TRANSCRIPTIONAL REGULATION NETWORKS
Claire Luciano
Department of Computer Science, UC San Diego, 9500 Gilman Drive, La Jolla, CA 92093, U.S.A.
Chun-Hsi Huang
Department of Computer Science and Engineering, University of Connecticut, Storrs, CT 06269, U.S.A.
Keywords: Network motif, Transcriptional regulation network, Sampling.
Abstract: Network motifs are small connected subnetworks within a larger network that occur in statistically
significant quantities and may indicate functional regions of the network. Network motif software tools
employ algorithms that compare a network to randomly generated networks in order to identify subnetworks
that occur in frequencies higher than would be expected by random chance. The transcriptional regulation
network of E. coli has been represented as a network and evaluated using both full enumeration and an edge
sampling algorithm. Several significant network motifs were identified, including feedforward loops and
bipartite graphs. This paper applies both full enumeration and a different sampling algorithm, randomized
enumeration, to the E. coli network using the newer software tool FANMOD. Evaluating the E. coli
transcriptional regulation network with FANMOD also identified feedforward loops and bipartite graphs as
significant network motifs. Sampling identified fewer and less significant motifs than full enumeration,
however, sampling enables the evaluation of larger subgraph sizes.
1 INTRODUCTION
Graph theory provides a useful mathematical model
to represent many systems as graphs composed of
vertices, or nodes, and edges that connect the pairs
of vertices. Network motifs are small connected
subnetworks within a larger network that occur in
higher frequencies than would be expected in
random networks (Kashtan 2004, Schreiber and
Schwobbermeyer 2005, Wernicke and Rasche
2006). Network motifs are the building blocks of
networks, providing information about the behavior
or design of the network; they may identify
functional regions within biological systems. A
range of network motif detection software tools have
been developed to analyze network systems and to
identify network motifs. Systems ranging from
social networks to biological systems have been
represented as graphs and analyzed for network
motifs using these software tools. However, there is
still room for improvement in network motif
detection software tools. Since the most common
network motifs in biological systems are different
than those found in other systems, it may be useful
to optimize software tools specifically for network
motif detection in biological systems. This could
improve both outcome and performance.
1.1 Network Motifs and E. Coli
The transcription regulation networks of E. coli and
Saccharomyces cerevisiae have been evaluated with
network motif detection software tools by Alon and
Lee respectively. There are three significant motif
patterns in the transcriptional regulation network of
E. coli. Each of these network motifs becomes
apparent while comparing subnetworks of a
particular size in the E. coli network with those of
the same size in randomly generated networks.
Significant motif patterns in E. coli depend on the
number of nodes within the system. For a graph
with only three nodes, the only
significant network
motif found by the Alon team is the feedforward
loop. The feedforward loop is characterized by three
nodes; X, Y and Z. A transcription factor X
regulates a second transcription factor Y, which both
187
Luciano C. and Huang C. (2010).
STRUCTURAL MOTIF ENUMERATION IN TRANSCRIPTIONAL REGULATION NETWORKS.
In Proceedings of the First International Conference on Bioinformatics, pages 187-192
DOI: 10.5220/0002760001870192
Copyright
c
SciTePress

jointly regulate the operon Z. Alon terms the X the
‘general transcription factor’, Y the ‘specific
transcription factor’ and Z the ‘effector operon’.
Feedforward loops are also significant network
motifs in networks with more than three nodes; in
this case, transcription factors X and Y jointly
regulate one or more operons Z(1)…Z(n). The
feedforward loop has other significant
characteristics, of which the most important is
coherence. Shen-Orr describes coherence, “A
feedforward loop motif is ‘coherent’ if the direct
effect of the general transcription factor on the
effector operons has the same sign (negative or
positive) as its net indirect effect through the
specific transcription factor. For example, if X and
Y both positively regulate Z, and X positively
regulates Y, the feedforward loop is coherent. If, on
the other hand, X represses Y, then the motif is
incoherent” (Shen-Orr, Shai, Milo, Mangan and
Alon, 2002). Most feedforward loops are coherent
(85%). The feedforward loop occurs much more
often within the E. coli transcriptional regulation
network than would be expected by random chance.
Shen-Orr suggests that the coherent feedforward
loop has a significant functional structure; the ability
to act as a circuit that rejects transient activation
signals from the general transcription factor and
responds only to persistent signals, while at the same
time allowing a rapid system shut down through the
control of the general transcription factor. This
structure is useful way to coordinate a rapid
response to an external signal. Also, the abundance
of coherent feedforward loops over incoherent loops
suggests a functional design. Lee’s research on the
transcriptional regulatory networks of
Saccharomyces cerevisiae suggest that the
feedforward loop is also a significant network motif
within that system (Lee et al., Science 02). This
suggests that the feedforward loop may also be
significant within other biological system networks.
Additional network motifs emerge as subgraphs of
increasing numbers of nodes are evaluated. When
subgraphs of four nodes are evaluated, the
overlapping regulation motif becomes apparent. In
the overlapping regulation motif, two operons are
regulated by the same two transcription factors.
This type of overlapping regulation motif is a
smaller and specific form of dense overlapping
regulons (DORs), which are discussed later.
Other significant motif patterns within the
transcriptional regulation network of E. coli can be
seen when graphs with higher numbers of nodes are
evaluated. When subgraphs of larger than three
nodes are evaluated, the single input module (SIM)
network motif becomes significant. The SIM is
defined by a set of operons that are controlled by a
single transcription factor, where all of the operons
are under control of the same sign (positive or
negative). There is no additional transcriptional
regulation of the operons. The transcription factors
involved in SIM systems are mostly autoregulatory
(70%). Most of the autoregulatory transcription
factors are autorepressive. There is a higher rate of
autoregulatory transcription factors within SIM
motifs than in the overall system. In the E. coli
transcription regulation network, 70% of the
transcription factors involved in SIM motifs are
autoregulatory, compared to 50% in the overall
dataset. SIMs are found in systems of genes that
function stochiometrically to form a protein
assembly (e.g. flagella) or a metabolic pathway (e.g.
amino acid biosynthesis). SIM systems may involve
temporal ordering, where the first gene activated is
the last to be deactivated.
Dense Overlapping Regulons (DORs) are a type
of network motif found within E. coli when
evaluating larger subnetworks. DORs are composed
of layers of overlapping interactions between
operons and a group of input transcription factors
organized in a bipartite graph that is much more
dense than corresponding structures in randomized
networks. DORs are not a homogenous mesh of
interconnections; rather, they contain several loosely
connected, internally dense regions of combinatorial
interactions. The regions are somewhat overlapping,
and different criteria can yield slightly different
groupings. One way to quantify DORs is by the
frequency of pairs of genes regulated by the same
two transcription factors. Shen-Orr uses a clustering
approach to define DORs. An algorithm detects
locally dense regions in the network with a high
ratio of connections to transcription factors. Within
the E. coli netw
ork, there are six DORs, where
operons in each DOR share common biological
functions. Usually, every output operon is
controlled by a different combination of input
transcription factors, but there are multi-input
modules in rare cases where several operons in a
DOR are regulated by precisely the same
combination of transcription factors with identical
regulation signs (termed ‘multi-input modules’.
DORs are significant in the larger structure of
biological networks; they seem to partition the
operons into biologically meaningful combinatorial
regulation clusters. DORs also govern how several
different network motifs connect together within the
larger network. Shen-Orr describes patterns in the
overall structure of the E. coli network, “A single
BIOINFORMATICS 2010 - International Conference on Bioinformatics
188

layer of DORs connects most of the transcription
factors to their effector operons. Feedforward loops
and SIMs often occur at the outputs of these DORs.
The DORs are interconnected by the global
transcription factors, which typically control many
genes in one DOR and a few genes in several
DORs” (Shen-Orr et al., Nature 02). Over 70% of
the operons are connected to DORs; the rest of the
operons are in small disjoint systems, with most
disjoint systems having only one to three operons.
1.2 Motif Detection Tools
There are a number of software tools dedicated to
network motif detection. Most of them employ
different algorithms to achieve this task. In order to
find network motifs, the software tool must find
which subgraphs occur in the input network and in
what number, determine which subgraphs are
isomorphic (equivalent), and determine which
subgraph classes of isomorphic graphs are displayed
at higher rates than in random graphs. This means
random graphs must also be generated. FANMOD
is a newer motif detection tool that uses a random
enumeration sampling algorithm. FANMOD uses
the NAUTY algorithm (McKay, 1981) in order to
group isomorphic graphs together into subgraph
classes. It also supports colored graphs, a useful
feature that other software tools do not support.
Support of colored graphs is a highly useful feature
for motif detection in biological networks because
elements that should not be connected to one
another, such as in a bipartite system, can be
assigned the same color. This is a computationally
effective way to avoid the generation of unnecessary
random graphs for comparison. FANMOD employs
a randomized enumeration algorithm called RAND-
ESU. It works by first taking an algorithm for full
enumeration and then modifying it to skip over some
subgraphs randomly as the algorithm is executed.
FANMOD also has the advantage of running much
faster than similar programs. Other software tools
include MAVISTO, which visualizes occurrences of
a motif in a network by a force-directed graph
algorithm, and MFINDER, which uses a different
algorithm called edge sampling (Wernicke and
Rasche, 2006). Edge sampling works by first
selecting a random edge in the input graph, and then
the edge is randomly extended until a connected
subgraph with the desired number of vertices is
obtained. However, edge sampling has distinct
disadvantages. Wernicke has shown that the edge
sampling algorithm results in a sampling bias, and
that the bias cannot be estimated from the number of
edges neighboring the oversampled subgraph alone.
1.3 This Study
Enumeration of the subgraphs of a particular size
within a larger graph is a computationally expensive
and time consuming task. As the size of the
subgraphs increases, the process becomes unwieldy
and current algorithms take far too long to execute.
Two of the major aspects involved in improving
network motif detection tools are improving full
enumeration algorithms for faster runtimes, and
improving the sampling of motifs so that the
algorithm is able to identify those motifs most likely
to be functionally relevant. The transcriptional
regulation network of E. coli has been analyzed by
Shen-Orr using a Markov-chain algorithm to
generate random networks for comparison.
FANMOD uses the previously discussed RAND-
ESU randomized enumeration algorithm to generate
random networks. We chose to evaluate Shen-Orr’s
E. coli transcriptional regulation network data with
FANMOD in order to see if other algorithms would
also identify the network motifs the Shen-Orr team
found significant.
The study consisted of analyzing Shen-Orr’s
E. coli transcription regulation network using the
FANMOD motif detection software tool. We
downloaded the E. coli transcription regulation
network data from the Uri Alon lab website to use as
the input file for FANMOD. Next, we ran both the
full enumeration and sampling algorithms with
FANMOD for subgraphs of increasing size, from
three nodes to five nodes. The sampling algorithm
takes less time to execute, but provides less
information and identifies fewer network motifs than
full enumeration. Sampling improves runtime, but
at the loss of the identification of functional network
motifs. The fact that the FANMOD sampling
algorithm returns far fewer significant network
motifs than full enumeration shows that there is
room to improve the sampling algorithm. However,
the more important quality in a sampling algorithm
is not so how many motifs it identifies compared to
full enumeration, but whether it is able to identify
those motifs that are most functionally relevant.
There are a few features that could improve the user
experience of FANMOD. FANMOD generates
diagrams of the most significant network motifs. It
would be useful to be able to highlight substructures
within these diagrams; for example, to highlight all
feedforward loops within subgraphs of a particular
size larger than three. Also, network motif detection
STRUCTURAL MOTIF ENUMERATION IN TRANSCRIPTIONAL REGULATION NETWORKS
189
tools could allow the user to specify the types of
network motifs that person is most interested in
seeing. For example, the ability to show graphs that
are bipartite or nearly so (80-90% of edges
connecting different colors) would be useful for
those studying such systems.
2 RESULTS
FANMOD provides several statistical values
alongside significant network motifs. The Z-score is
one way of determining how significant a network
motif is. The Z-Score is the original frequency
minus the random frequency divided by the standard
deviation. Motifs with the highest Z-scores are the
most significant, so the following tables of motifs
are organized in order of decreasing Z-score. P-
Values range from zero to one; smaller p-Values
indicate more significant motifs because a smaller p-
value indicates that the motif occurs more often in
the network than would occur by random chance.
The p-Value is calculated in the following way,
“The p-Value of a motif is the number of random
networks in which it occurred more often than in the
original network, divided by the total number of
random networks.”
The following tables show the results for full
enumeration and sampling of the E. coli network,
enumerating subgraphs of size three. All graphs are
ordered by descending Z-Score, so that the most
significant network motifs are listed first. Full
enumeration of the network at subgraph size three
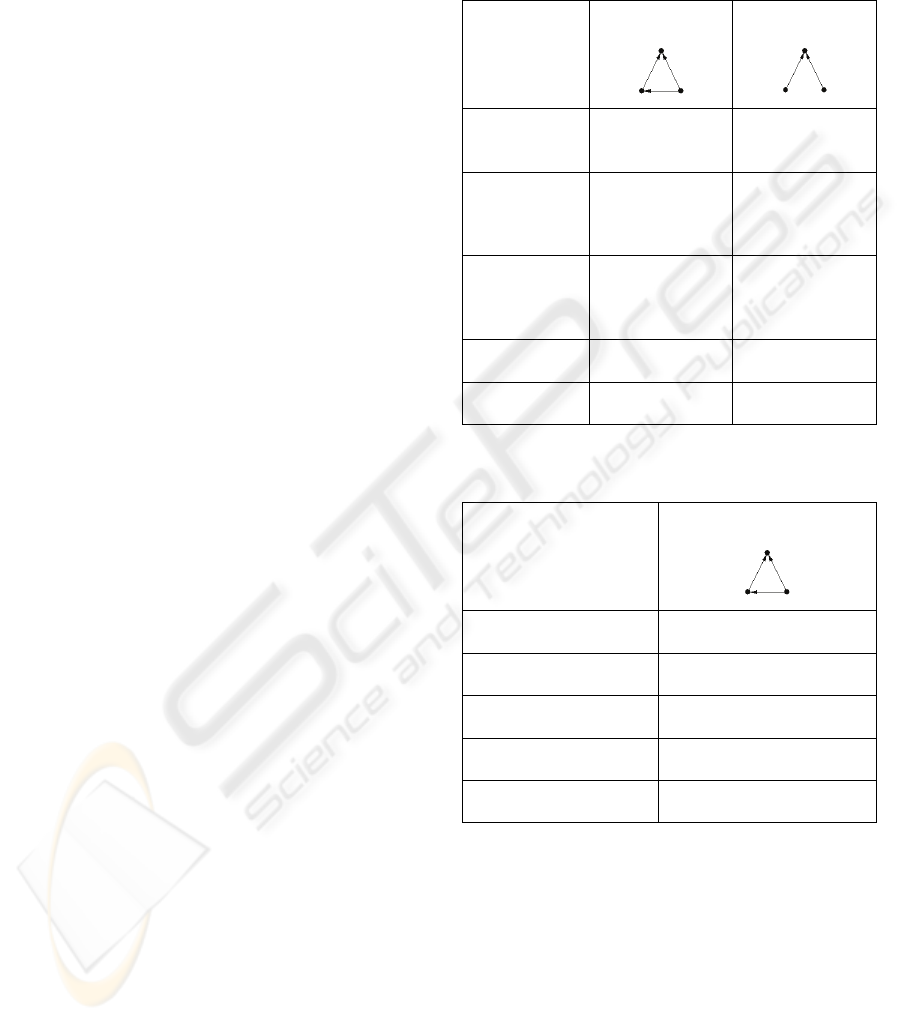
identified two significant network motifs, whereas
the sampling algorithm just identified one significant
network motif. The full enumeration data shows the
average values from three trials. Two of the five
trials for the sampling algorithm of
subgraph size
three resulted in no identification of significant
network motifs. The sampling data shows the
average values from the remaining three trials.
The table below shows the three most significant
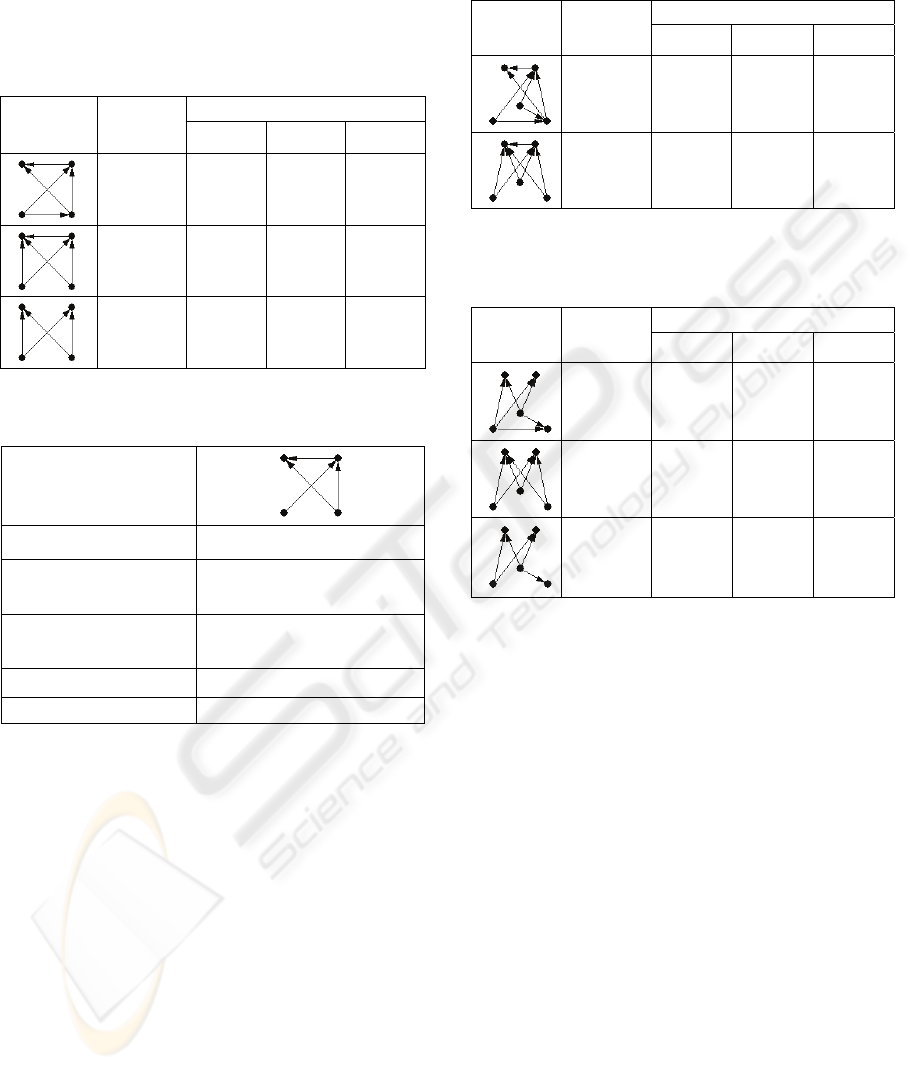
network motifs of subgraph size four identified
using full enumeration. All of the seven significant
network motifs for subgraph size four using full
enumeration are bipartite graphs or contain at least
one feedforward loop. The two motifs with the
highest Z-scores contain two feedforward loops, and
the third most significant is a bipartite graph. The
remaining four motifs contain one feedforward loop.
For sampling of subgraph size four, only one trial
out of three trials identified any significant network
motifs at all. The one network motif identified was
the sixth most significant according to the full
enumeration data.
Table 1: Significant network motifs identified using
FANMOD, full enumeration and subgraph size 3 (n=3).
(Three trials)
Feedforward
Loop
Bipartite
Graph
Average
Frequency
[Original]
0.80676% 91.76%
Average
Mean-
Frequency
[Random]
0.14468%
91.216%
Average
Standard-
Deviation
[Random]
0.00058784 0.00048442
Average
Z-Score
11.264 11.221
Average
p-Value
0 0
Table 2: Significant network motifs identified using
FANMOD, sampling and subgraph size 3 (n=3).
(Three Trials)
Feedforward
Loop
Average Frequency
[Original]
0.99259%
Average Mean-
Frequency [Random]
0.10969%
Average Standard-
Deviation [Random]
0.0015703
Average Z-Score 5.6127
Average p-Value 0.003667
The full enumeration algorithm identified the 20
most significant network motifs for subgraphs of
size five. Each of the three trials identified 20
network motifs; 22 distinct motifs total. All of the
22 motifs are either bipartite graphs or contain at
least one feedforward loop. Two of the 22 network
motifs contain three feedforward loops and three of
the motifs are bipartite graphs. Ten of the motifs
contain two feedforward loops, and the remaining
seven graphs contain only one feedforward loop
each. The table below shows the two network
motifs found with full enumeration of subgraph size
five that contain three feedfoward loops. The first
BIOINFORMATICS 2010 - International Conference on Bioinformatics
190
network motif listed was found to have the highest
Z-score for all three trials, and the second was in the
top ten motifs in every trial. The next table shows
the three bipartite graphs.
Table 3: Significant network motifs identified using
FANMOD, full enumeration and subgraph size 4 (n=4).
Network
Motif
Average
Z-score
Motif Rank
Trial 1 Trial 2 Trial 3
24.927 1 1 1
15.641
2 2 2
10.533
3 3 3
Table 4: Significant network motifs identified using
FANMOD, sampling and subgraph size 4 (n=4).
Network Motif
Frequency [Original] 0.62167%
Mean-Frequency
[Random]
0.050375%
Standard-Deviation
[Random]
0.00074301
Z-Score 7.6889
p-Value 0.001
The sampling algorithm identified five
significant motifs in one trial and ten in another.
The third trial identified no significant motifs. The
two trials together identified 12 distinct motifs. 9 of
these 12 motifs are included in the 22 motifs
identified using full enumeration. Of the 12 motifs
identified using sampling, one motif contains three
feedforward loops, one motif contains two
feedforward loops, and seven contain one
feedforward loop. The remaining three motifs
contain no feedforward loops. The second motif
containing three feedforward loops in the table
below was also identified in one of the sampling
trials, ranked first with a Z-score of 10.528. As can
be seen in the table below, the average Z-score for
the same motif identified using full enumeration is
16.047.
Table 5: Significant network motifs containing three
feedforward loops, identified full enumeration and
subgraph size 5 (n=5).
Network
Motif
Average
Z-score
Motif Rank
Trial 1 Trial 2 Trial 3
206.22 1 1 1
16.047 6 8 9
Table 6: Significant network motifs that are bipartite
graphs, identified full enumeration and subgraph size 5
(n=5).
Network
Motif
Average
Z-score
Motif Rank
Trial 1 Trial 2 Trial 3
17.474 7 7 6
11.31933
10 11 10
5.6591
18 19 17
3 CONCLUSIONS
Every single significant network motif identified
using full enumeration for subgraph sizes of three,
four and five is either a bipartite graph or contains
one or more feedforward loops. Most of the
network motifs identified using sampling also
contain one or more feedforward loops. The data
supports the notion that the feedforward loop and
bipartite graphs are statistically significant structures
in the transcriptional regulation network of E. coli.
The data supports Shen-Orr’s research that shows
the feedforward loop as the most significant network
motif at subgraph size three. Additionally, the
second network motif that was detected using full
enumeration is a bipartite graph, suggesting that
bipartite graphs are significant in the E. coli
transcription regulation network. Many of the
network motifs found to be significant from the
enumeration and sampling of larger subnetwork
sizes contain feedforward loops or are bipartite
graphs. When the subgraph size is increased to four,
the full enumeration data shows that the two most
STRUCTURAL MOTIF ENUMERATION IN TRANSCRIPTIONAL REGULATION NETWORKS
191

significant network motifs each contain two
feedforward loops. The third most significant motif
is the previously-described overlapping regulon
motif, a bipartite graph. All of the remaining
identified motifs for full enumeration of size four
graphs contain one feedforward loop with one
additional edge. Overall, six out of the seven
significant motifs contain at least one feedforward
motif, with the two most significant motifs
containing two feedforward loops. The remaining
motif is a bipartite graph. This further supports that
feedforward loops and bipartite systems are
significant within the E. coli transcription regulation
network. The sampling data for subgraphs of size
four only identified one of the significant motifs, the
sixth most significant according to the full
enumeration data. It consists of a feedforward loop
with one additional edge. The most significant
motifs at subgraph size five also contain feedforward
loops, with those ranked highest containing two or
three feedforward loops. The sampling data showed
fewer network motifs than full enumeration in all
cases. For subgraphs of sizes four and five, the most
significant motif identified by sampling was not
represented in the top five most significant motifs
identified using full enumeration. Sampling has
major shortcomings when compared to full
enumerations; it identifies both fewer and less
significant network motifs. Overall, the FANMOD
data shows that feedforward loops and bipartite
graphs are significant network motifs in the
transcriptional regulation network of E. coli.
4 FUTURE WORK
There are several ways to improve network motif
detection tools. One way to improve sampling
results in FANMOD could be to alter the default
probability settings in order to better favor sampling
the network more evenly. This is discussed in detail
in Section 5 of the FANMOD manual. The default
settings are 0.5 for all probability fields, but
organizing the probability fields with high
probabilities in the left fields and lower probabilities
in the right fields increases the chance that the
network will be sampled more evenly. However,
preliminary results for sampling using the
probabilities 0.8, 0.5 and 0.3 for subgraph size three
resulted in identification of the feedforward loop
with a lower average Z-score from three trials
(4.8304) than sampling subgraphs of size three with
the probabilities 0.5, 0.5, 0.5 (Z-score 5.6127). This
indicates that the feedforward loop was actually
found to be less significant using the alternative
descending probabilities. More trials could be
conducted using subgraphs of different sizes to
determine whether an altered probability pattern
improves the results from sampling compared to
those from full enumeration.
FANMOD also supports several models for
randomized network generation. Our study used the
local constant model, where directed edges are
exchanged with one another and the number of
edges connected to each vertex remains constant.
FANMOD also supports a global constant model
that preserves the total number of edges, but the
number of edges connected to a specific vertex may
or may not remain the same. The transcriptional
regulation network of E. coli could also be evaluated
using the global constant model with full
enumeration and sampling in order to see whether
the motifs identified as significant change.
Additionally, it would be useful to test other
transcriptional regulation networks to see if
structures such as the feedforward loop are
significant in other biological networks.
REFERENCES
Kashtan, N., Itzkovitz, K., Milo, R. and Alon, U. (2004)
“Efficient sampling algorithm for estimating subgraph
concentrations and detecting network motifs”,
Bioinformatics, 20(11):1746-1758.
Lee, Tong Ihn et al. (2002) “Transcriptional Regulatory
Networks in Saccharomyces Cerevisiae,” Science,
298(5594):799-804.
McKay, B. (1981) “Practical Graph Isomorphism”,
Congressus Numerantium, Vol 30, 45-87.
Milo, R., Shen-Orr, S. Itzkovitz, K., Chklovskii, D.
and Alon, U. (2002) “Network Motifs: Simple
Building Blocks of Complex Networks”, Science,
298(5594):824-827.
Schreiber. F. and Schwobbermeyer, H. (2005)
“Mavisto: a tool for the exploration of network
motifs,” Bioinformatics, 21(17), 3572-3574.
Shen-Orr, Shai S., Milo, R., Mangan, S. and Alon, U.
(2002) “Network motifs in the transcriptional
regulation network of Escherichia coli,” Nature
Genetics, Vol 31, 64-68.
Wernicke, S. and Rasche, F. (2006) “FANMOD: a tool
for fast network motif detection,” Bioinformatics,
22(9):1152–1153.
BIOINFORMATICS 2010 - International Conference on Bioinformatics
192
