
PROTEIN SECONDARY STRUCTURE PREDICTION USING
KNOWLEDGE-BASED POTENTIALS
Saras Saraswathi, Robert L. Jernigan, Andrzej Kloczkowski
Department of Biochemistry, Biophysics and Molecular Biology
L.H. Baker Center for Bioinformatics and Biological Statistics
112 Office and Laboratory Building, Ames, IA, 50011, U.S.A.
Andrzej Kolinski
Laboratory of Theory of Biopolymers, Faculty of Chemistry, Warsaw University, Pasteura 1, 02-093 Warsaw, Poland
Keywords: Protein secondary structure prediction, Neural networks, Extreme learning machine, Particle swarm
optimization.
Abstract: A novel method is proposed for predicting protein secondary structure using data derived from knowledge
based potentials and Neural Networks. Potential energies for amino acid sequences in proteins are
calculated using protein structures. An Extreme Learning Machine classifier (ELM-PSO) is used to model
and predict protein secondary structures. Classifier performance is maximized using the Particle Swarm
Optimization algorithm. Preliminary results show improved results.
1 INTRODUCTION
Large scale advances in genome sequencing and
resultant availability of large numbers of proteins
sequences has given protein secondary structure
prediction increasing importance in computational
biology. Improvements in secondary structure
prediction can lead to progress in protein
engineering and drug design. Existing
crystallographic techniques are too expensive and
time consuming for large-scale determination of
protein three-dimensional structures. Prediction of
secondary structures might be a useful intermediate
step to speed up structure prediction (Lomize,
Pogozheva and Mosberg, 1999 and Ortiz, Kolinski,
Rotkiewicz, Ilkowski and J. Skolnick, 1999).
Secondary structure prediction can assist in gene
function and sequence annotation, as well as
identification and classification of structures and
functional motifs and in identifying malfunctioning
structures which cause human diseases.
Several computational methods have been
successfully used in secondary structure prediction,
of which empirical and machine learning methods
have proved to be the most successful. Chou and
Fasman (1974), Qian and Sejnowski (1988), Ward,
McGuffin, Buxton, and Jones (2003) were followed
by numerous others. The GOR method based on
information theory was used by Garnier,
Osguthorpe, and B. Robson, (1978) and later by
Garnier, Gibrat, and Robson (1996). Kloczkowski,
K.L. Ting, R.L. Jernigan, and J. Garnier (2002) used
evolutionary information in GOR V for improved
structure prediction. PredictProtein server (Rost, G.
Yachdav, and J. Liu, 2004) uses multiple sequence
alignment based neural networks. The PSIPRED
algorithm developed by Jones (1999) uses PSI-
BLAST (Altschul, T.L. Madden, A.A. Schäffer, J.
Zhang, Z. Zhang, W. Miller, and D.J. Lipman, 1997)
and neural networks. The Jpred prediction server
(Cole, Barber, and Barton, 2008), runs on the Jnet
algorithm (Cuff and Barton, 2000). Montgomerie,
Sundaraj, Gallin, and Wishart (2006) and Pollastri,
Martin, Mooney and Vullo, (2007) developed large
scale secondary structure prediction methods using
existing structural information and computational
methods to claim an accuracy of 90% for sequences
with over 30% sequence homology. Kihara (2005)
suggested that long-range interactions are an
important factor to be considered in order to achieve
higher classification accuracy.
370
Saraswathi S., Jernigan R., Kloczkowski A. and Kolinski A..
PROTEIN SECONDARY STRUCTURE PREDICTION USING KNOWLEDGE-BASED POTENTIALS.
DOI: 10.5220/0003086903700375
In Proceedings of the International Conference on Fuzzy Computation and 2nd International Conference on Neural Computation (ICNC-2010), pages
370-375
ISBN: 978-989-8425-32-4
Copyright
c
2010 SCITEPRESS (Science and Technology Publications, Lda.)

We propose a novel strategy for secondary
structure prediction using knowledge based potential
profiles. A two stage Extreme Learning Machine
(ELM) (Huang, Q.Y. Zhu, and C.K. Siew, 2006)
classifier called the ELM-PSO is used for
classification of secondary structures. Performance
is improved using Particle Swarm Optimization
(PSO) (Clerc, J. kennedy, 2002).
This paper is organized as follows: Section 2
gives a brief description of the data. Section 3
describes the two-stage ELM-PSO classification
technique. Section 4 discusses the results and gives a
comparative study followed by conclusions in
Section 5.
2 DATA GENERATION USING
POTENTIAL ENERGY
The dictionary of secondary structure assignment
Database of Secondary Structure in Proteins (DSSP)
(Kabsch and Sander, 1983) has 8 classes of protein
secondary structures. We use only a reduced set of
three secondary structures, namely, alpha-helix (H),
beta-strand (E) and coil (C). Data is derived based
on CABS force-fields (Kolinski, 2004 – algorithm
for data generation has been submitted for
publication), which includes information pertaining
to long and short range interactions between amino
acids in proteins. A profile matrix was created using
the 513 non-homologous (target) protein sequences
from the CB513 data set (Cuff and Barton, 2000),
where the sequence homology is less than 30%.
3 METHODS
AND OPTIMIZATION
In a neural net framework, the input consists of a set
of patterns (residues), each having a set of 27
features (profile values), which are normalized to
values between 0 and 1. The output consists of three
units which correspond to one of three secondary
structure elements, represented as a 1 for the class of
interest and a -1 for the other two classes. A given
input is combined with a bias and a set of weights
and is processed through an activation function at
the hidden layer level. The output of the hidden
layer is combined with another set of weights to
yield three outputs. The predicted class is considered
as the output which has the maximum value, which
corresponds to choosing the output with the smallest
mean-squared error.
An Extreme Learning Machine (ELM) (
Huang,
Zhu, and Siew, 2006) classifier, which is a form of a
Neural Network, is used for classification. PSO
is
used to tune the parameters of the ELM. The data
was also evaluated using Support Vector Machine
(SVM) and Naïve Bayes (NB) algorithms using the
WEKA (
Witten and Frank,2005) software tool for
classification.
The profile data consists of 27 features for each
of N amino acids, where N is the number of residues
in a single protein. Of the 27 features, the first 9
features are the energy potentials related to alpha-
helices (H), the next 9 features are related to beta-
strands (E) and the last 9 features are related to coils
(C) as seen in Fig. 1 and 2. This gives a particular
advantage in getting better classification accuracy,
since this information can be used during the
training phase (although this information will not be
available on a blind set or a new set of proteins).
Based on this prior knowledge, class specific
features of the target class can be given extra
weights (importance) compared to the rest of the
features that belong to the negative classes. Hence
the class specific features of each class (9 columns
per class) were scaled (values boosted) according to
a predetermined factor prior to building a training
model. These factors (not unique) were obtained by
brute force trial and error method, where selection
was based on getting better classification results. It
is noteworthy that the classification accuracy after
this scaling depends on the scaling factors used, and
ranges from 60% (for non-scaled data or data scaled
with sub-optimal boosting values), to over 95%,
when the optimal scaling factors are used. The first 9
features of all samples belonging to the H class,
were scaled by a factor of 5, while the second set of
9 features were scaled by a factor of 3 and the last
set of 9 features were scaled by a factor of 8. The
scaling of data improves the classification accuracy
considerably during the training phase. Samples
which were scaled according to their classes were
used for the 10-fold cross-validation in WEKA
(
Witten and Frank, 2005), which gave very high results for
SVM and Naïve Bayes algorithms. Since it is not
possible to perform class-specific feature scaling
during testing (blind) phase for the ELM method,
three sets of test samples were generated for each
sample in the test set. The first set had the first 9
features boosted in the same ratio as for the H class
for all samples. The second set of test samples had
the next set of 9 features boosted according to the
factor used for the E class for all samples and the
third set of test samples had the last set of 9 features
scaled according to the factor used for the C class
PROTEIN SECONDARY STRUCTURE PREDICTION USING KNOWLEDGE-BASED POTENTIALS
371

for all samples. Each test set was sent in turn and the
votes were collected for the classification. For
robustness, ten sets of training models were used to
get the classification results for the same test set.
Each training model yielded a set of three votes for
each sample. These votes were all gathered to
determine the class which receives the maximum
number of votes. The results for the classification
accuracies with and without feature scaling (value
boosting) are given in the results section. Blind
testing with voting was not done for SVM and Naïve
Bayes algorithms since it would require
modification of WEKA code.
Figure 1: Visualization of data without feature scaling.
Energy potentials are represented along the x-axis, the first
nine features belong to helix (H), the next 9 features are
for strand (E) and the last set of features 19 - 27 for coil
(C). The color intensity indicates the value of the potential
energy, with a dark blue for a low value and a red
indicates a high value. The residues (total: 4282) along the
y-axis have been sorted according to the three classes,
where residues 1 - 1487 belong to class H, 1488 - 2541
belong to class E and 2542 - 4282 belong to class C. Note:
there is not much horizontal differentiation among the
three classes which becomes evident in Fig. 2, after data is
subjected to feature specific scaling. Results for
classification of this unscaled data is given in Table 1.
3.1 Two Stage Extreme Learning
Machine
The ELM-PSO consists of the Extreme Learning
Machine (ELM) classifier as the main algorithm,
which uses a set of training samples to build a
model. During the training phase, PSO is called
upon to optimize the parameters, such as weights,
number of hidden neurons and bias of the ELM,
which results in improved classification accuracy.
These parameters are stored and used during the
testing phase. ELM is an improved version of a
feed-forward neural network consisting of a single
hidden layer. The initial set of input weights are
chosen randomly, but they are tuned later by the
PSO. The output weights from the hidden layer to
the output layer are analytically calculated, using a
pseudo inverse. A sigmoidal activation function is
used for the hidden layer and a linear activation
function is used for the output neurons. Huang,
Zhu
and Siew
(2006) give a comprehensive discussion of
ELM. The ELM algorithm consists of the following
steps:
1. Select the number of hidden neurons (H) and a
suitable activation function for a given problem.
2. Randomly choose the input weight (W) and bias
(b).
3. Analytically calculate the output weight using a
pseudo inverse which speeds up the traditional
neural network algorithm tremendously.
4.
Store the calculated weights (W, b) and hidden
neurons (H) which yield the best training results.
5.
Use these stored values for estimating the class
label during testing phase.
The estimated class label Ci is calculated using
equation (1) where
k
i
y
is the neural network output
for each class k, for sample i.
k
i
Ck
i
yc
,..,2,1
maxarg
ˆ
(1)
An improved version of the ELM algorithm
proposed by Saraswathi and Suresh et al., (2010),
shows that a random selection of initial parameters
(W, b, H) affects the performance of the ELM
classifier significantly. Tuning of input parameters
using PSO, improves classifier performance
considerably, by minimizing the error (Eq. 2), which
is the distance between the neural network output
(Y) and the target classes (T).
TYminargb,W,H
b,V,H
***
(2)
3.2 Particle Swarm Optimization
A stochastic optimization technique called Particle
Swarm Optimization (PSO) was developed by
Clerc
and Kennedy (2002).
This method mimics the
intelligent social behavior of flocks of birds or
schools of fish, represented as particles in a
population. These particles work together to find a
simple and optimal solution to a problem in the
shortest possible time. The PSO algorithm is
initialized with a set of random solutions called
particles. The algorithm iteratively searches a multi-
ICFC 2010 - International Conference on Fuzzy Computation
372

dimensional space for the best possible solution,
determined by a fitness criterion. PSO will find the
best combination of hidden neurons, input weights,
and bias values and return the (training) validation
efficiency obtained by the ELM algorithm along
with the best ELM parameters to obtain better
generalization performance. The best parameters
are stored and used during the testing phase.
4 RESULTS AND DISCUSSION
Several training models were built using ELM and
two other algorithms, namely SVM and Naïve Bayes
(NB) from the WEKA (
Witten and E. Frank, 2005) suit
of software for data classification. A 10-fold cross
validation was performed for SVM and NB, where
90% of the proteins were used to build the training
model while the remaining 10% were retained for
testing the model, but all input information was
scaled according to previously described values. A
blind test was conducted using ELM with 4797
proteins for training and 4835 for testing. These
residues were selected from a random selection of
30 proteins for the training set out of 400 proteins,
while the test samples came from a separate set of
41 proteins retained for testing. Preliminary studies
for the ELM-PSO classifier, SVM and NB show
high accuracies of around 99% for the scaled
training as seen in Table 2, while the results for the
unscaled version of the data, as seen in Table 1, is
much lower at only ~60% or less. The unscaled
version of the data uses only row specific feature
information while the scaled data also uses column
specific class information which increases the
accuracy considerably.
The lower testing accuracy of 94.4 % for the
ELM (blind) tested
4835 samples might be due to
the smaller number of residues tested as compared to
the other two models built from SVM and NB with
the full data set. The ELM classifier trains on sets of
2000 to 3000 samples at a time and builds several of
these models by selecting samples at random from
the pool of available training samples (from the 400
training proteins), a very computationally intensive
process. The parameters for every ELM model are
optimized by calling PSO and a single pattern from
the test set is repeatedly tested by each model,
giving a consensus classification for the type of the
test sample. The class that occurs with the highest
frequency in these classifications is taken to be the
predicted class for this test sample. Preliminary
results for a set of 4835 test samples are given in
Table1 and Table 2 for scaled and unscaled data.
On the other hand the high accuracies for SVM
and NB can be attributed to the technique of cross
validation where the input data is uniformly scaled
according to previous criteria, using feature specific
class information, which results in higher accuracy.
There is no blind test of data. So, unless the
algorithm can discern this feature specific pattern
automatically without involving the computationally
intensive ELM-PSO method that was used here, it is
not very practical. Future work will aim to improve
the ELM-PSO algorithm to learn this information
automatically.
Table 3 shows that the ELM-PSO methods
perform very well compared to other studies in the
literature for scaled data. The accuracy on the
unscaled data is lower for all models and is
comparatively low for the blind test, indicating that
the learning algorithm needs further tuning to
discern the column-wise information during (blind)
testing phase. The column-wise class information is
a unique feature of our data that separates the three
classes linearly and hence gives high results. Table1
and Table 2 also give the F-measure and area under
the curve (AUC) values for SVM and Naïve Bayes
classifications. These terms help us to gauge the
quality of the predictions.
Figure 2: Visualization of the same sample data shown in
Figure 1, given here with feature scaling. Descriptions of
the X,Y axes and colors are the same as given in Figure 1.
Compared to Figure 1, it can be seen that class-specific
feature scaling provides for a distinct separation of the
classes, which results in higher accuracy during
classification, using ELM, SVM-SMO and Naïve Bayes
algorithms, with results shown in Table 2.
The performance of classifications can be
evaluated in terms of the true positives (TP-correct)
and false positive (FP-error) terms. Similar
definition holds for true negatives (TN) and false
negatives (FN). The output of a classification might
provide estimated probabilities which determine the
predicted class according to a pre-set threshold. TP
rate and FP rate can be graphed as coordinate pairs
PROTEIN SECONDARY STRUCTURE PREDICTION USING KNOWLEDGE-BASED POTENTIALS
373
which form the receiver operating characteristic
curve (ROC curve).
The area under this ROC curve (AUC or
AUROC) helps to aggregate the performance of all
the testing results, where a higher value closer to
1.00 denotes perfect performance. F-measure gives
the test’s accuracy. It uses precision p and recall r of
the test, where p is the ratio of correct results divided
by all returned results (TP/(TP+FP)) and r is the
number of correct results divided by the number of
expected results (TP/(TP+FN)). F-measure is
calculated as given in equation (3), where the best
score for F-measure can be as high as 1 and the
worst score can be as low as 0.
recallprecision
)recall*precision(*2
measure_F
(3)
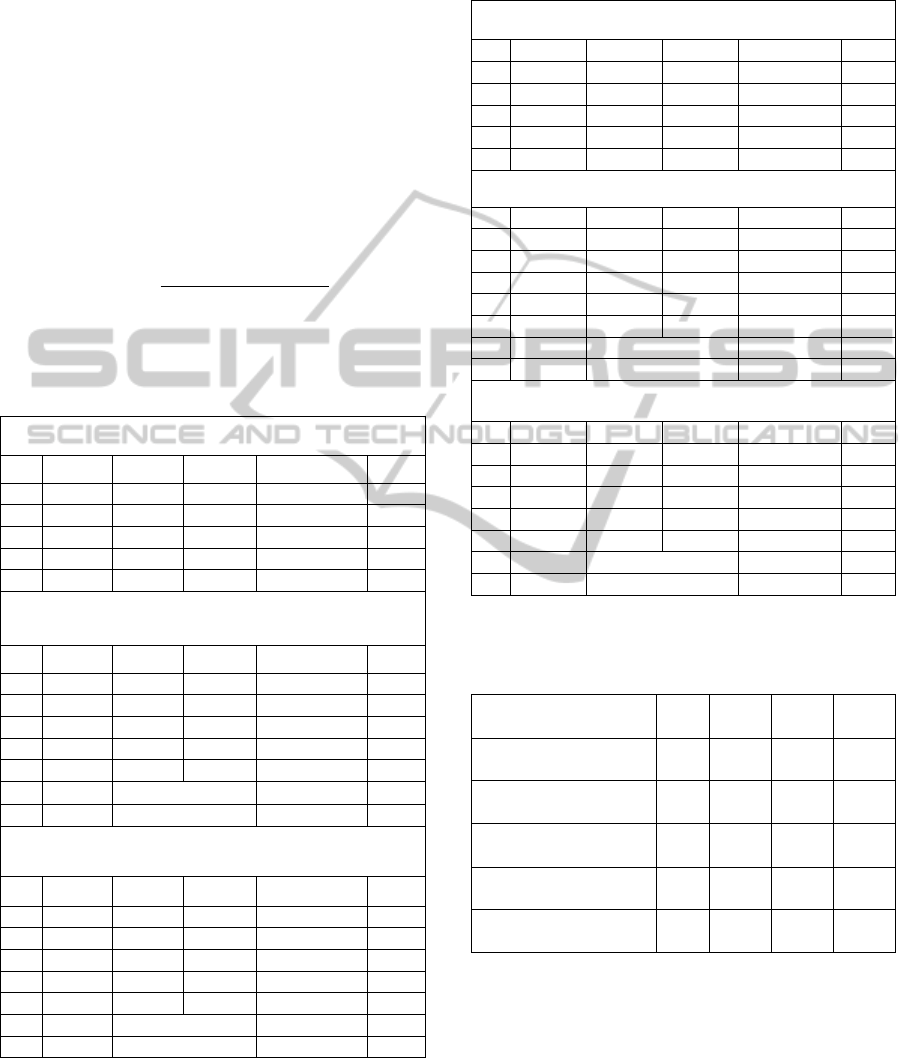
Table 1: Confusion matrix and accuracies for the three
classes of secondary structures, for data without feature
scaling, using ELM-PSO, SVM and Naïve Bayes.
Confusion Matrix – ELM-PSO – without feature scaling
H E C % correct
H 1147 116 457 66.7 QH
E 300 329 474 27.1 QE
C 604 175 1195 30.6 QC
Total 4797
55.7 Q3
Confusion Matrix – SVM – without feature scaling
H E C % correct
H 3153 533 1672 58.8 QH
E 817 1353 1411 22.8 QE
C 1446 595 5083 20.5 QC
Total 16063
59.7 Q3
F-Measure
58.5
AUC
70.0
Confusion Matrix – Naïve Bayes – No feature scaling
H E C % correct
H 3244 1217 897 60 QH
E 705 2168 708 60 QE
C 2028 2168 708 47.6 QC
Total 16063
54.8 Q3
F-Measure 55.1
AUC 73.5
Table 2: Confusion matrix for the three classes of
secondary structures, for data with feature scaling, using
ELM-PSO, SVM and Naïve Bayes.
Confusion Matrix – ELM-PSO – with feature scaling
H E C % correct
H 1814 0 0 100 QH
E 56 942 94.3 QE
C 224 0 1799 89.9 QC
Total 4835
94.4 Q3
Confusion Matrix – SVM - with feature scaling
H E C % correct
H 24854 67 8 99.7 QH
E 0 16879 4 100 QE
C 0 0 31096 100 QC
Total 72908
99.9 Q3
F- Measure 99.8
AUC 99.9
Confusion Matrix – Naïve Bayes - with feature scaling
H E C % correct
H 24896 33 0 99.9 QH
E 256 16627 0 98.5 QE
C 0 19 31077 99.9 QC
Total 72908
99.6 Q3
F- Measure 99.6
AUC 100
Table 3: Comparison of results for secondary structure
prediction using ELM-PSO - feature scaled data, with
other studies in literature.
Method
Q3
( %)
QH
(%)
QE
(%)
QC
(%)
PHD (Rost and
Sander, 1999)
70.8 72.2 66.0 72.0
JNet server (Cuff and
Barton, 2000)
76.4 78.4 63.9 80.6
SVMpsi (Kim and
Park, 2003)
76.6 78.1 65.6 81.1
SPINE server (Dor
and Zhou, 2007)
80.0 84.44 72.23 80.46
ELM-PSO with
feature scaling
94.4
100 94.3 89.9
5 CONCLUSIONS
A two stage approach for secondary structure
prediction was presented where an Extreme
Learning Machine (neural network) was used along
with Particle Swarm Optimization (ELM-PSO) for
classifying a reduced set of three secondary
ICFC 2010 - International Conference on Fuzzy Computation
374

structures, namely, alpha-helix, beta-strand and coil.
The data was generated using CABS potential
energy. ELM-PSO needs improvement to achieve
better accuracies on blind tests so that comparative
results can be achieved on new proteins.
ACKNOWLEDGEMENTS
We thank Pawel Gniewk, a student at ‘Theory of
Biopolymers, Faculty of Chemistry, Warsaw
University’, whose original idea and algorithm was
used to generate the potentials data that was used for
the secondary structure predictions. We
acknowledge the support of National Institutes of
Health through grants R01GM081680,
R01GM072014, and R01GM073095 and the support
of the NSF grant through IGERT-0504304.
REFERENCES
Altschul, S., Madden, T., and Schaffer, A., 1997. Gapped
BLAST and PSI-BLAST: a new generation of protein
database search programs. Nucl Acids Res, 25, 3389 –
3402.
Clerc, M. K. and Kennedy, J., 2002. The particle swarm -
explosion, stability, and convergence in a
multidimensional complex space. IEEE Trans
olutionary Comput, 6 (1) 58-73.
Chou, P. Y. and Fasman, G. D., 1974. Prediction of protein
conformation. Biochemistry, 13(2), 222-245.
Cole, C., Barber, J. D., and Barton , G. J., 2008. The Jpred
3 secondary structure prediction server. Nucleic Acids
Research, 36 (Web Server issue): W197–W201.
Cuff, J. A. and Barton, G. J., 2000. Application of multiple
sequence alignment profiles to improve protein
secondary structure prediction. Proteins, 40(3), 502-
511.
Dor, O. and Zhou, Y., 2007. Achieving 80% Ten-fold
Cross-validated Accuracy for Secondary Structure
Prediction by Large-scale Training. PROTEINS:
Structure, Function, and Bioinformatics, 66, 838-845.
Garnier, J., Osguthorpe, D. J. and Robson, B., 1978.
Analysis of the accuracy and implications of simple
methods for predicting the secondary structure of
globular proteins. J Mol Biol, 1, 97-120.
Garnier, J., Gibrat, J. F., and Robson, B., 1996. GOR
secondary structure prediction method version IV.
Methods Enzymol, 226, 540-553.
Huang, G. B., Zhu, Q. Y., and Siew, C. K., 2006. Extreme
learning machine: Theory and applications.
Neurocomputing, 70(1-3), 489-501.
Jones, D., 1999. Protein secondary structure prediction
based on position-specific scoring matrices. J Mol
Biol, 292, 195 – 202.
Kabsch, W. and Sander, C., 1983. Dictionary of protein
secondary structure: pattern recognition of hydrogen-
bonded and geometrical features. Biopolymers, 22(12),
2577-2637.
Kihara, D., 2005. The effect of long-range interactions on
the secondary structure formation of proteins. Prot
Sci., 14( 8), 1955–1963.
Kim, H. and Park, H., 2003. Protein Secondary Structure
Prediction Based on an Improved Support Vector
Machines Approach. Protein Eng, 16, 553-560.
Kloczkowski, A., Ting, K. L., Jernigan, R. L., and Garnier,
J., 2002. Combining the GOR V algorithm with
evolutionary information for protein secondary
structure prediction from amino acid sequence.
Proteins, 49, 154-166.
Kolinski A., 2004. Protein modeling and structure
prediction with a reduced representation. Acta Biochim
Pol, 51, 349-371.
Lomize, A. L., Pogozheva, I. D. and Mosberg, H. I., 1999.
Prediction of protein structure : The problem of fold
multiplicity. Proteins, 37, 199-203.
Montgomerie, S., Sundaraj, S., Gallin W., and Wishart, D.,
2006. Improving the Accuracy of Protein Secondary
Structure Prediction Using Structural Alignment. BMC
Bioinformatics, 7, 301.
Ortiz,
A. R., Kolinski, A., Rotkiewicz, P., Ilkowski, B. and
Skolnick, J., 1999. Ab initio folding of proteins using
restraints derived from evolutionary information.
Proteins Suppl 3 (CASP3 Proceedings), 177-185.
Pollastri, G., Martin, A., Mooney, C. and Vullo, A., 2007.
Accurate prediction of protein secondary structure and
solvent accessibility by consensus combiners of
sequence and structure information. BMC
Bioinformatics, 8(1), 201.
Qian, N. and Sejnowski, T. J., 1988. Predicting the
secondary structure of globular proteins using neural
network models. J Mol Biol, 202, 865-884.
Rost, B. and Sander, C., 1993. Prediction of protein
secondary structure at better than 70% accuracy. J.
Mol. Biol., 232, 584–599.
Rost, B., 2001. Review: Protein Secondary Structure
Prediction Continues to Rise. J Struct Bio, 134, (2-3),
204-218.
Rost, B., Yachdav, G. and Liu, J., 2004. The PredictProtein
Server, Nucl Acids Res, 32, Web Server issue, W321-
W326.
Saraswathi, S., Suresh, S., Sundararajan, N., Zimmerman,
M. and Nilsen-Hamilton, M., 2010. ICGA-PSO-ELM
approach for Accurate Cancer Classification Resulting
in Reduced Gene Sets Involved in Cellular Interface
with the Microenvironment. IEEE Transactions in
Bioinformatics and Computational Biology,
http://www.computer.org/portal/web/csdl/doi/10.1109/
TCBB.2010.13.
Ward, J. J., McGuffin, L. J., Buxton, B. F. and Jones, D.
T., 2003. Secondary structure prediction with support
vector machines. Bioinformatics, 19(13), 1650-1655.
Witten, I. H. and Frank, E., 2005. Data Mining: Practical
machine learning tools and techniques, (2nd ed.) San
Francisco: Morgan Kaufmann.
PROTEIN SECONDARY STRUCTURE PREDICTION USING KNOWLEDGE-BASED POTENTIALS
375
