
RECOGNITION OF GENE/PROTEIN NAMES USING
CONDITIONAL RANDOM FIELDS
David Campos, S
´
ergio Matos and Jos
´
e Lu
´
ıs Oliveira
Institute of Electronics and Telematics Engineering of Aveiro, University of Aveiro
Campus Universit
´
ario de Santiago, 3810-193 Aveiro, Portugal
Keywords:
Natural Language Processing, Text Mining, Machine Learning, Named Entity Recognition, Gene/Protein
Names.
Abstract:
With the overwhelming amount of publicly available data in the biomedical field, traditional tasks performed
by expert database annotators rapidly became hard and very expensive. This situation led to the development
of computerized systems to extract information in a structured manner. The first step of such systems requires
the identification of named entities (e.g. gene/protein names), a task called Named Entity Recognition (NER).
Much of the current research to tackle this problem is based on Machine Learning (ML) techniques, which
demand careful and sensitive definition of the several used methods. This article presents a NER system using
Conditional Random Fields (CRFs) as the machine learning technique, combining the best techniques recently
described in the literature. The proposed system uses biomedical knowledge and a large set of orthographic and
morphological features. An F-measure of 0,7936 was obtained on the BioCreative II Gene Mention corpus,
achieving a significantly better performance than similar baseline systems.
1 INTRODUCTION
In the last decades, there was an explosion of the pub-
licly available data, a consequence of the deep inte-
gration of computerized solutions in society. This
overwhelming amount of textual information was also
verified in biomedicine, with the rapid growth in the
number of published documents, such as articles,
books and technical reports. MEDLINE (Medical
Literature Analysis and Retrieval System Online) is
the U.S. National Library of Medicine (NLM) pre-
mier bibliographic database, and it contains over 19
million references to journal papers in life sciences.
It continues to be daily updated, and since 2005,
between 2000-4000 completed references are added
each day (National Center for Biotechnology Infor-
mation, 2009). MEDLINE and other biomedical re-
sources, such as GenBank, PIR, and Swiss-Prot are
manually curated by expert annotators, in order to
correctly identify biological entities (e.g., proteins,
genes, and pathways) on texts, organizing the ex-
tracted information in a structured format. However,
with the large amounts of data, this becomes a hard
and very expensive task. This situation naturally led
to the development of computerized systems, which
perform various automated techniques such as named
entity recognition and relationship extraction.
Information Extraction (IE) is the task of extract-
ing instances of predefined categories from unstruc-
tured data (e.g., natural language texts), building a
structured and unambiguous representation of the en-
tities and the relations between them (Franz
´
en et al.,
2002). One of the research areas of IE is Named
Entity Recognition (NER), which involves process-
ing structured and unstructured documents and iden-
tifying expressions that refer to entities of interest.
For instance, on the identification of entities such as
persons, locations and e-mail addresses from texts.
There are several solutions to implement automated
NER systems, including rule-based, dictionary-based,
machine learning and hybrid approaches. This arti-
cle will focus on Machine Learning (ML) techniques,
which use methods to learn how to recognize specific
entity names. The learning procedure uses texts con-
taining entity names annotated by experts. This ap-
proach solves some of the dictionary-based problems,
recognizing new spelling variations of an entity name.
However, ML does not provide direct ID information
of recognized entities, such as GenBank ID or Swis-
sProt ID, which can be solved using a dictionary in an
extra step.
This field of research has received considerable at-
275
Campos D., Matos S. and Oliveira J..
RECOGNITION OF GENE/PROTEIN NAMES USING CONDITIONAL RANDOM FIELDS.
DOI: 10.5220/0003096902750280
In Proceedings of the International Conference on Knowledge Discovery and Information Retrieval (KDIR-2010), pages 275-280
ISBN: 978-989-8425-28-7
Copyright
c
2010 SCITEPRESS (Science and Technology Publications, Lda.)

tention in recent years, and many systems have been
developed, using distinct techniques to reach the same
goal. The main characteristics and differences be-
tween the several systems will be presented. The
first characteristic relies on the ML technique, varying
between semi-supervised and supervised methods.
Semi-supervised systems combine unlabelled and la-
belled data, such as in the work presented by (Ando,
2007). On the other hand, supervised learning tech-
niques use only labelled data to train a model. There
is more research in this technique, and consequently
a panoply of research works using different models,
such as Conditional Random Fields (CRFs), Hidden
Markov Models (HMMs), Support Vector Machines
(SVMs) and Maximum Entropy Models (MEMMs).
In addition to the ML technique, it is common to
combine distinct models in one system. For instance,
trough the combination of a model trained reading
the text forward with other reading backward (Ando,
2007), or by using two or more models using different
ML techniques (Huang et al., 2007). The second char-
acteristic relies on the type of features applied in the
machine learning technique. Orthographic, morpho-
logical and Part of Speech (POS) features are com-
monly used. A system presented by (Vlachos, 2007)
extends this idea, using a syntactic parser to generate
multiple POS tags for each token to mitigate unseen
errors. The output of this parser makes it possible to
establish relations between tokens within a sentence
independently of their proximity. Finally, it is also
common to use domain specific concepts as features,
performing matching between text and large lexicons
(Chen et al., 2007). The final characteristic is the us-
age of post-processing techniques, in order to filter
and correct errors generated by the recognition step.
The most common used methods are abbreviation res-
olution, dictionary filtering and parenthesis matching.
In this article we present a system to extract
gene/protein names from biomedical documents, de-
scribing the used methods and comparing the results
with existent systems with equivalent characteristics.
2 METHODS
In a text mining problem, it is necessary to train a
model based on natural language texts. However, it is
necessary to define strategies to extract features from
text, and use those features to define the chunks of
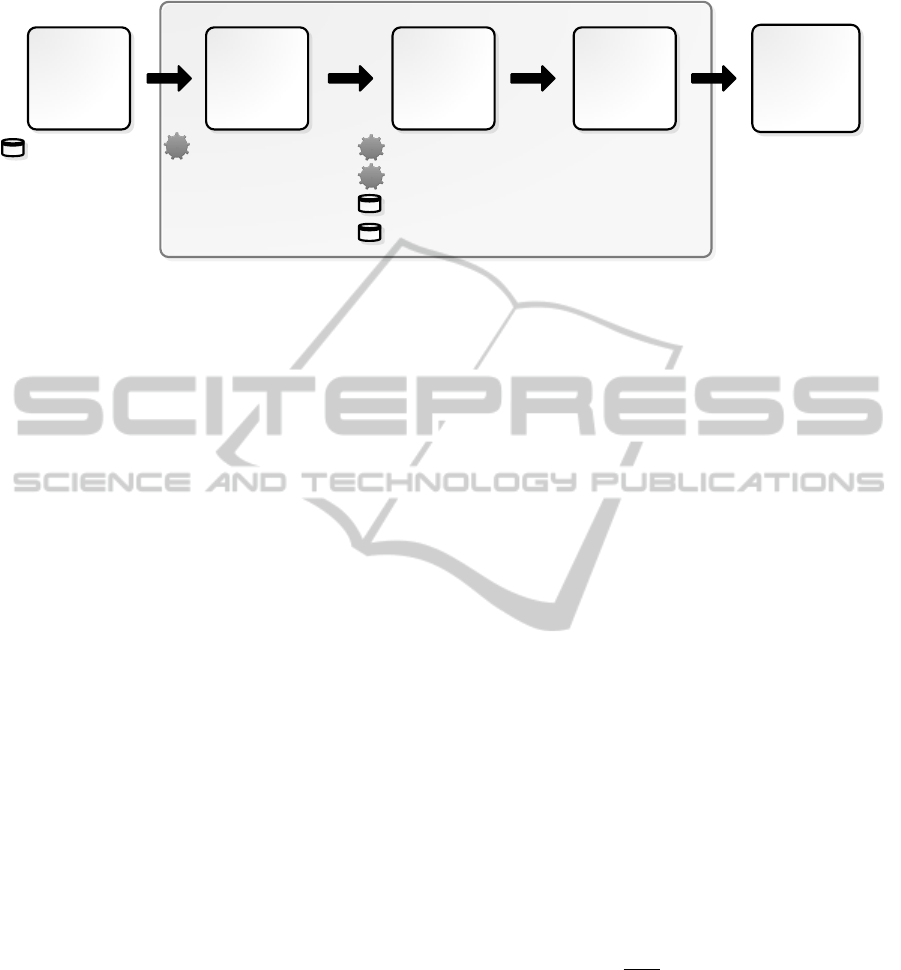
text that are gene/protein names. Figure 1 presents the
system’s architecture, focusing on the pipeline and on
the several used tools and resources.
2.1 Corpus
The first step is to obtain a set of texts to train and test
the implemented system. In order to train the model
to recognize entity names with the highest accuracy as
possible, all gene/protein names must be precisely an-
notated by human experts. There are several corpora
publicly available, such as BioCreative, GENIA (Kim
et al., 2003), and BioNLP (Johnson et al., 2007). In
this work, the BioCreative II corpus for Named Entity
Recognition (Smith et al., 2008) is used. It is part of
the BioCreative challenge, which is an international
competition for NER, Normalization and detection of
protein-protein interactions. It is composed of 15000
sentences for training and 5000 sentences for testing,
and contains 44500 annotations of Human gene/pro-
tein names.
2.2 Tokenization
In order for NER tasks to be accomplished by com-
puterized systems in an effective manner, it is neces-
sary to divide natural language texts into meaningful
units, called tokens. A token is a group of characters
that is categorized according to a set of rules.
The tokenization process is one of the most im-
portant tasks of the whole workflow, since all the fol-
lowing tasks will be based on tokens resulting from
this process. A technical report from the National Li-
brary of Medicine (He and Kayaalp, 2006) debates
the performance of several existent tokenizers. The
main goal of this work was to find a tokenizer that re-
turns tokens with a minimum loss of information for
MEDLINE articles, exposing the advantages and lim-
itations of the several available solutions. The cho-
sen tokenizer highly depends on the user’s require-
ments. In this document the authors concluded that
the OpenNLP (Baldridge et al., 2010) and SPECIAL-
IST NLP (Browne et al., 2000) tokenizers break a
given text into small pieces by delimiting both at
white spaces and punctuations, respecting the defined
requirements. OpenNLP was the chosen tokenizer
for this system for two main reasons: a) it preserves
hyphenated compound words and various numerical
forms within a single token boundary, which is very
common on gene/protein names; and b) it is a train-
able tokenizer, which allows to train it with a cus-
tomized training set or apply the syntax model pro-
vided by default.
2.3 Features
The features are the input of the machine learning
method, which will use them to predict if a specific
KDIR 2010 - International Conference on Knowledge Discovery and Information Retrieval
276
Feature
Extraction
CRF
Annotated
Gene/Protein
Names
OpenNLP POS Tagger
BioThesaurus
BioLexicon
Snowball Stemmer
Tokenization
OpenNLP Tokenizer
Corpus
BioCreative II GM
MALLET
Figure 1: Global workflow of the system, specifying the external tools and/or resources used on each step.
chunk of text is an entity name or not. In text min-
ing it is necessary to extract these features from texts.
This process requires special attention, because it is
necessary to define a wide set of features that will re-
flect the special phenomena and linguistic character-
istics of the naming conventions. The final goal is to
identify only the necessary features, removing those
that do not contribute to an increase of performance.
Based on the experience of previous works and
after various tests, we obtained the best set of fea-
tures (Table 1), which reaches the system’s peak per-
formance.
2.4 Model
In order to identify if each token is part of an entity
name or not, it is necessary to use an encoding method
that assigns a tag to each word of the text. These tags
will be used as classes by the classifiers. There are
several techniques to accomplish this goal, such as IO,
BIO, BMEWO and BMEWO+. Our system uses the
BIO approach, which is the de facto standard. It uses
the tag “B” to identify the tokens that are the begin-
ning of an entity name, tag “I” to identify the tokens
that are the continuation of the name, and tag “O” to
the tokens that are outside of any entity name.
There are several solutions to find a model in
order to predict the class of each token. Our sys-
tem uses CRFs, because they have several advantages
over other methods. At first, CRFs avoid the label
bias problem (Lafferty et al., 2001), a weakness of
MEMMs. On the other hand, CRFs also have ad-
vantage over HMMs, a consequence of its conditional
nature, which results in the relaxation of the indepen-
dence assumptions, in order to ensure tractable infer-
ence. CRFs outperformed both MEMMs and HMMs
on a number of real-world sequence labeling tasks
(Lafferty et al., 2001). Regarding SVMs, a indepth
study (Keerthi and Sundararajan, 2007) showed that
when the two methods are compared using identical
feature functions they do turn out to have quite close
peak performance. However, SVMs may take a large
amount of time to generate even the simplest models.
2.4.1 Conditional Random Fields
Conditional Random Fields were first introduced by
Lafferty et al. (Lafferty et al., 2001). Assuming that
we have an input sequence of observations (repre-
sented by X), and a state variable that needs to be
inferred from the given observations (represented by
Y ), a CRF is a form of undirected graphical model
that defines a single log-linear distribution over label
sequences (Y ) given a particular observation sequence
(X).
This layout makes it possible to have efficient al-
gorithms to train models, in order to learn conditional
distributions between Y
j
and feature functions from
training data. To accomplish this, it is necessary to
determine the probability of a given label sequence
Y given X, and consequently the most likely label.
At first, the model assigns a numerical weight to each
feature, then those weights are combined to determine
the probability of a certain value for Y
j
. This proba-
bility is calculated as follows:
p(y|x, λ) =
1
Z(x)
exp(
∑
j
λ
j
F
j
(y, x)), (1)
where λ
j
is a parameter to be estimated from training
data and indicates the informativeness of the respec-
tive feature, Z(x) a normalization factor and F
j
(y, x)
the sum of state or transition functions that describe a
feature.
In this work, we use the CRFs’ implementation of
MALLET (McCallum, 2002), a Java-based package
for statistical natural language processing, document
classification, clustering, topic modelling and infor-
mation extraction.
RECOGNITION OF GENE/PROTEIN NAMES USING CONDITIONAL RANDOM FIELDS
277

Table 1: Complete set of machine learning features used by our system.
Feature Description Resources/Tools
Token and Stem Use Token and its Stem to group together the different in-
flected forms of a word.
Snowball Stemmer (Porter,
2001)
Part of Speech Marking up the words in a text as corresponding to a partic-
ular grammatical category.
OpenNLP POS Tagger
(Baldridge et al., 2010)
Orthographic Capture knowledge about token’s formation. Regular expressions
Morphological Locate common structures and/or subsequences of charac-
ters between several entity names.
Regular expressions
Special Symbols Tag Greek words and Roman Digits. -
Dictionary Matching Match dictionary gene/protein entries with the natural lan-
guage text.
BioThesaurus (Liu et al., 2006)
Relevant Concepts Mark domain specific concepts that indicate the presence of
entity names.
Dictionary of domain terms
(e.g. nucleobases, nucleosides,
amino acids and DNA/RNA se-
quences)
Relevant Verbs Tag verbs that could indicate the presence of entity names in
the surrounding tokens.
BioLexicon (Sasaki et al.,
2008)
Window Model local context using a -1,1 window of features. -
3 RESULTS AND DISCUSSION
In order to evaluate the system’s accuracy, it is nec-
essary to calculate measures that provide precise and
global feedback about its behaviour. To obtain those
measures, each prediction must be classified as True
Positive (TP), True Negative (TN), False Positive (FP)
or False Negative (FN). Using this strategy, it is pos-
sible to calculate the ability of the system to present
only relevant items (P-Precision) and to present all
relevant items (R-Recall). The overall system perfor-
mance is usually measured in terms of the F-measure
(F), calculated as the harmonic mean of precision and
recall. Those measures are calculated as follows:
P =
T P
T P + FP
, R =
T P
T P + FN
,
F = 2 ×
P ×R
P +R
In order to compare the presented system with pre-
vious works, we have selected the systems from the
BioCreative II Gene Mention Task that are more sim-
ilar to our implementation. Thus, only systems that
use one CRF, without combining it with other ma-
chine learning techniques or dictionary lookup, were
considered.
The first system, presented on (Grover et al.,
2007), applies a series of linguistic pre-processing
methods, including tokenization, lemmatization, part
of speech tagging, chunking and abbreviation detec-
tion. The chunker creates structural information that
includes words of the text, recognizing boundaries of
simple noun and verb groups. This system also uses
dictionary matching, concepts from the biomedical
domain and head nouns (determined by the chunker)
as features.
The second system presented by (Vlachos, 2007)
uses a wide set of features, including the token itself,
information about whether it contains digits, letters or
punctuation, capitalization, and also prefixes and suf-
fixes. In addition, it extracts more features from the
output of a syntactic parser, which generates multi-
ple POS tags for each token in order to mitigate un-
seen token errors. The syntactic parser output is in the
form of grammatical relations, which can link tokens
within a sentence independently of their proximity.
The system presented on (Tsai et al., 2006) uses
seven feature types: word, bracket, orthographical,
part of speech, character n-grams and dictionary
matching. It also performs a post-processing task, us-
ing global patterns composed of gene mention tags
and surrounding words to refine the recognition pro-
cess.
Finally, the system presented by (Sun et al., 2007)
uses orthographical, context, word shape, prefix and
suffix, part of speech, and shallow syntactic features.
It does not use any specific domain features.
Table 2 lists the results and characteristics of the
several systems. The performance of our system is
above the average (F-measure of 0.7859) of the sys-
tems that participated on the BioCreative II Gene
Mention Task, where a large part of them use an en-
semble of classifiers or a combination with dictionary
lookup. In one case, our system outperforms a system
that combines two SVMs (Chen et al., 2007).
Regarding the systems that use only one CRF, the
two top systems implement a strategy to extract con-
text knowledge (chunking and syntactic parsing), es-
KDIR 2010 - International Conference on Knowledge Discovery and Information Retrieval
278

Table 2: Comparison of our system with selected systems from the BioCreative II Gene Mention Task.
System Precision Recall F-measure Characteristics
(Grover et al., 2007) 0.8697 0.8255 0.8470 CRF + Abbreviation Detection + Chunker
(Vlachos, 2007) 0.8628 0.7966 0.8284 CRF + Syntactic Parsing - Domain Concepts
Our 0.8796 0.7227 0.7936 CRF
(Tsai et al., 2006) 0.9267 0.6891 0.7905 CRF + Post Processing
(Sun et al., 2007) 0.8046 0.7361 0.7688 CRF - Domain Concepts
tablishing relations between the several tokens in a
sentence. In our system, the relations are limited to a
{-1,1} window, because it reached the best results in
comparison with bigger windows. However, we ex-
tract less contextual information, which has showed
to be crucial to better recognize multi-token gene/pro-
tein names. This observation raises the importance of
using as much contextual information as possible.
Considering systems that do not relate all the to-
kens of the sentence, our system outperforms the oth-
ers, even when post-processing methods are used.
Overall, our system is the third best when using only
one CRF model.
From this comparison, the performance results
showed that contextual features have more impact
than post processing methods and specific domain
concepts. Our system achieves better results than the
system presented by (Tsai et al., 2006), which uses
a post processing technique to refine the recognized
names. On the other hand, the system presented by
(Vlachos, 2007) has better results than our system
without using domain concepts.
4 CONCLUSIONS
In this paper we presented a system to recognize
gene/protein names from natural language texts, using
Conditional Random Fields as the machine learning
technique. A large set of orthographic and morpho-
logical features is used, in order to extract precise and
complete knowledge about words’ shape. Dictionary
matching and specific domain concepts are also used
as features, in order to improve the overall system’s
recall. Compared to other systems that use weak con-
textual information, our system reached best results,
reaching an F-measure of 0.7936.
From the analysis of our results and the compar-
ison to other similar systems, it seems that explor-
ing more gene/protein names databases, in order to
match more names correctly and consequently in-
crease the impact of the dictionary matching feature,
could be beneficial. Another important point is the
introduction of more domain specific concepts. For
instance, UMLS terminology could be used to help
on gene/protein names recognition. Moreover, the in-
tegration of more features could also explored, trying
to extract more morphological and orthographic in-
formation (e.g., word length). We also intend to ex-
plore techniques to collect more contextual informa-
tion, which showed to have a strong contribution to
performance, both on recall and precision. Finally, in
order to increase the performance of the implemented
system, distinct models may be combined, taking ad-
vantage of the different predictions provided by each
model on the same chunk of text.
ACKNOWLEDGEMENTS
D. Campos is funded by Fundac¸
˜
ao para a Ci
ˆ
encia
e Tecnologia (FCT) under the project PTDC/EIA-
CCO/100541/2008. S. Matos is funded by FCT under
the Ci
ˆ
encia2007 programme.
REFERENCES
Ando, R. (2007). BioCreative II gene mention tagging sys-
tem at IBM Watson. In Proceedings of the Second
BioCreative Challenge Evaluation Workshop, pages
101–103. Citeseer.
Baldridge, J., Morton, T., and Bierner, G. (2010). openNLP
Package.
Browne, A. C., McCray, A. T., and Srinivasan, S. (2000).
The SPECIALIST LEXICON. Technical report, Lis-
ter Hill National Center for Biomedical Communica-
tions, National Library of Medicine.
Chen, Y., Liu, F., and Manderick, B. (2007). Gene men-
tion recognition using lexicon match based two-layer
support vector machines. In Proceedings of the Sec-
ond BioCreative Challenge Evaluation Workshop; 23
to 25 April 2007; Madrid, Spain.
Franz
´
en, K., Eriksson, G., Olsson, F., Asker, L., Lid
´
en, P.,
and C
¨
oster, J. (2002). Protein names and how to find
them. Int J Med Inform, 67(1-3):49–61.
Grover, C., Haddow, B., Klein, E., Matthews, M., Nielsen,
L., Tobin, R., and Wang, X. (2007). Adapting a
relation extraction pipeline for the BioCreAtIvE II
task. In Proceedings of the second BioCreative chal-
lenge evaluation workshop, volume 23, pages 273–
286. Citeseer.
RECOGNITION OF GENE/PROTEIN NAMES USING CONDITIONAL RANDOM FIELDS
279

He, Y. and Kayaalp, M. (2006). A Comparison of 13 To-
kenizers on MEDLINE. Technical report, The Lister
Hill National Center for Biomedical Communications.
Huang, H., Lin, Y., Lin, K., Kuo, C., Chang, Y., Yang,
B., Chung, I., and Hsu, C. (2007). High-recall gene
mention recognition by unification of multiple back-
ward parsing models. In Proceedings of the Second
BioCreative Challenge Evaluation Workshop, pages
109–111. Citeseer.
Johnson, H., Baumgartner, W., Krallinger, M., Cohen, K.,
and Hunter, L. (2007). Corpus refactoring: a feasibil-
ity study. Journal of biomedical discovery and collab-
oration, 2(1):4.
Keerthi, S. and Sundararajan, S. (2007). CRF versus SVM-
Struct for sequence labeling. Technical report, Yahoo
Research.
Kim, J., Ohta, T., Tateisi, Y., and Tsujii, J. (2003). GE-
NIA corpus-a semantically annotated corpus for bio-
textmining. Bioinformatics-Oxford, 19(1):180–182.
Lafferty, J., McCallum, A., and Pereira, F. (2001). Con-
ditional random fields: Probabilistic models for seg-
menting and labeling sequence data. In Proceedings of
the Eighteenth International Conference on Machine
Learning (ICML-2001). Citeseer.
Liu, H., Hu, Z.-Z., Zhang, J., and Wu, C. H. (2006). Bio-
thesaurus: a web-based thesaurus of protein and gene
names. Bioinformatics, 22(1):103–105.
McCallum, A. K. (2002). MALLET: A Machine Learning
for Language Toolkit. http://mallet.cs.umass.edu/.
National Center for Biotechnology Information (2009).
Medline fact sheet.
Porter, M. (2001). Snowball: A language for stemming al-
gorithms.
Sasaki, Y., Montemagni, S., Pezik, P., Rebholz-Schuhmann,
D., McNaught, J., and Ananiadou, S. (2008). Biolex-
icon: A lexical resource for the biology domain. In
Proc. of the Third International Symposium on Se-
mantic Mining in Biomedicine (SMBM 2008), vol-
ume 3.
Smith, L., Tanabe, L., Ando, R., Kuo, C., Chung, I., Hsu,
C., Lin, Y., Klinger, R., Friedrich, C., Ganchev, K.,
et al. (2008). Overview of BioCreative II gene men-
tion recognition. Genome biology, 9(Suppl 2):S2.
Sun, C., Lei, L., and Xiaolong, W. and, Y. G. (2007).
A study for application of discriminative models in
biomedical literature mining. In Proceedings of the
Second BioCreative Challenge Evaluation Workshop;
23 to 25 April 2007; Madrid, Spain.
Tsai, R., Sung, C., Dai, H., Hung, H., Sung, T., and Hsu, W.
(2006). NERBio: using selected word conjunctions,
term normalization, and global patterns to improve
biomedical named entity recognition. BMC bioinfor-
matics, 7(Suppl 5):S11.
Vlachos, A. (2007). Tackling the BioCreative2 gene men-
tion task with conditional random fields and syntac-
tic parsing. In Proceedings of the Second BioCreative
Challenge Evaluation Workshop; 23 to 25 April 2007;
Madrid, Spain, pages 85–87. Citeseer.
KDIR 2010 - International Conference on Knowledge Discovery and Information Retrieval
280
