
A METHOD TO IMPROVE THE ACCURACY OF PROTEIN
TORSION ANGLES
J. C. Calvo, J. Ortega, M. Anguita
Department of Computer Architecture and Computer Technology, CITIC-UGR, University of Granada, Granada, Spain
J. Taheri, A. Zomaya
School of Information Technologies, University of Sydney, Sydney, Australia
Keywords:
Proteins, Torsion angles, Rotamer libraries.
Abstract:
Protein structure prediction (PSP) is an open problem with many useful applications in disciplines such as
Medicine, Biology and Biochemistry. As this problem presents a vast search space where the analysis of
each protein structure requires a significant amount of computing time, it is necessary to propose efficient
search procedures in this very large space of possible protein conformations. Thus, an important issue is
to add vital information (such as rotamers) to the process to decrease its active search space –rotamers give
statistical information about torsional angles and conformations. In this paper, we propose a new method
to refine the torsional angles of a protein to remake/reconstruct its structures with more resemblance to its
original structure. This approach could be used to improve the accuracy of the rotamer libraries and/or to
extract information from the Protein Data Bank to facilitate solution of the PSP problem.
1 INTRODUCTION
Proteins have important biological functions such as
the enzymatic activity of the cell, attacking diseases,
transport and biological signal transduction, among
others. They are chains of amino acids whose se-
quences determine their 3D structure after a folding
process. Moreover, as in almost all cases, the func-
tionality of proteins is exclusively determined by their
corresponding 3D structure, there is a high interest in
knowing a reasonably accurate 3D structure for any
given protein.
The experimental determination of the 3D struc-
ture of a protein using methods such as X-ray crys-
tallography and nuclear magnetic resonance (NMR)
is usually complex and costly. As a result, less
than a 0.6% of the protein sequences included in
UniProt (UniProt, 2008) have a known structure in the
PDB (Protein Data Bank) (Zhang and Skolnick, 2005;
RCSB, 2009). Toward solving this problem, an alter-
native approach, called Protein Structure Prediction
(PSP), is used to take advantage of present comput-
ing capabilities to determine/predict the 3D structure
of a protein given its sequence of amino acids (Lesk,
2002).
There are 20 different amino acids. Each amino
acid can be divided into two main areas: backbone
and side-chain. All amino acids have the same back-
bone, but different side-chains to individualize them.
A protein is a chain of amino acids where the junction
between amino acids is provided by their backbones.
Figure 1 shows a sample protein with its amino acids’
backbones and side-chains.
Torsion angles
H
C
H
H
C
C
N
O
CH
C
C
H
H
H
H
H
C
HH
H
Backbone
Side-chain
N
φ
ω
ψ
χ
1
χ
2
χ
3
χ
4
Figure 1: Torsion angles in a sample amino acid.
To store attributes and characteristics of
proteins, it is necessary for them to be effi-
297
C. Calvo J., Ortega J., Anguita M., Taheri J. and Zomaya A..
A METHOD TO IMPROVE THE ACCURACY OF PROTEIN TORSION ANGLES.
DOI: 10.5220/0003137502970300
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2011), pages 297-300
ISBN: 978-989-8425-36-2
Copyright
c
2011 SCITEPRESS (Science and Technology Publications, Lda.)
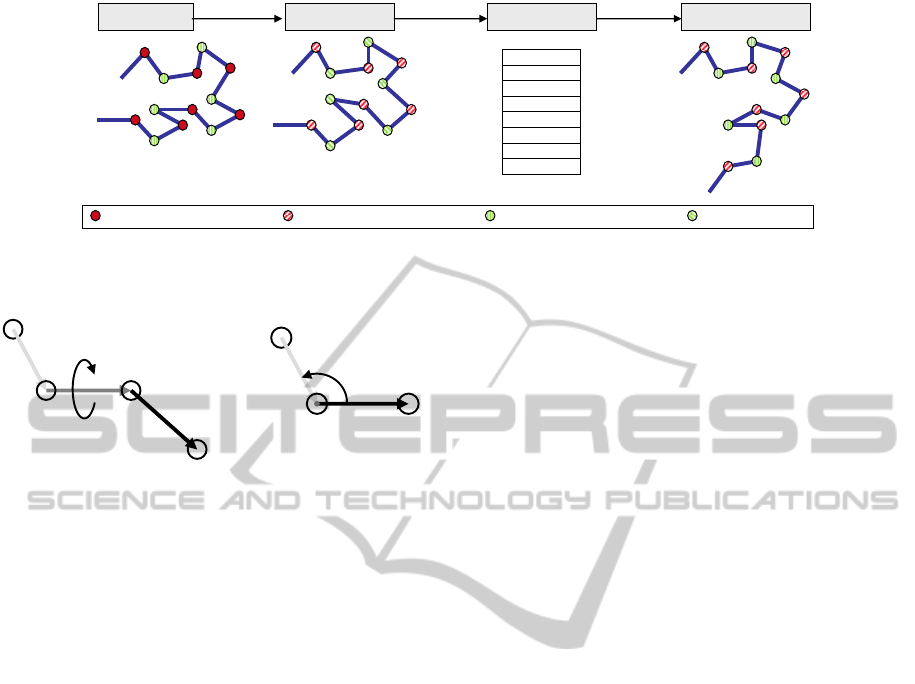
Noisy torsion angle Real static angle
Real torsion angle Noisy static angle
Real protein Noisy protein Remade protein
X-Ray
Torsion
Angles
extraction
PDB file
Torsion angles
3.24
2.45
56.13
-120.56
-75.25
170.10
17.90
…
Remaking
a) b) c) d)
Noisy torsion angle Real static angle
Real torsion angle Noisy static angle
Real protein Noisy protein Remade protein
X-Ray
Torsion
Angles
extraction
PDB file
Torsion angles
3.24
2.45
56.13
-120.56
-75.25
170.10
17.90
…
Optimization
Process
a) b) c) d)
Optimized Torsion angles
3.11
2.41
54.13
-121.22
-72.45
170.47
16.90
…
e)
Remaking
Figure 3: (a) A real protein’s structure, (b) its PDB structure with noticeable noise in atom positions, (c) torsion angles
extracted from the PDB, (d) remade protein with very different structure because of cumulative noises.
a
b
c
a
b
c
Figure 2: Representation of a torsion angle in the bond b
from two points of view.
ciently/mathematically represented. All-atom 3D
coordinates, main-atom 3D coordinates, backbone
atoms coordinates and side-chain centroids, and
torsion angles are typical approached deployed for
this purpose. As a general rule, representations
based on 3D coordinates have the common problem
of not always being able to reconstruct feasible
proteins based on their restored 3D information. In
contrast, torsion angles can always represent valid
protein conformations when correct bond lengths
and static angles are available or assumed. Hence,
torsion angles are mainly used to reconstruct and
represent proteins. In this case, each amino acid has
3 torsion angles in the backbone (φ, ψ and ω) and a
variable number of torsion angles in the side-chain
(0 to 4 depending on the amino acid). Therefore,
for a medium-size protein with 60 amino acids, the
number of torsion angles can vary between 180 and
420.
The Protein Data Bank contains all known pro-
tein structures obtained by traditional procedures such
as X-Ray and NMR. Although these methods are as-
sumed to obtain/calculate proteins’ structures with
RMSD (Root Mean Square Deviation) of around 2
˚
A –depending on the size of the protein–, PDB files
always have some level of noise in their 3D coordi-
nates. Although such noise affects all atoms of a pro-
tein, overall shape of the constructed protein is usu-
ally fairly similar to the real protein. Figure 2 repre-
sents a torsion angle between three atom bonds a, b,
and c; and, equation 1 demonstrate how such torsion
angle is mathematically calculated. In this equation:
a, b and c are vectors in ℜ
3
, ‘×’ is the vectorial prod-
uct, ‘·’ is the dot product, and atan2 computes arc
tangent with two parameters and returns the principal
value of the arc tangent of y/x in radians.
φ = atan2(|b|a ·[b × c], [a × b]· [b ×c]) (1)
Although it seems fairly easy to reconstruct a pro-
tein based on its torsion angles, the affecting noises
in these torsion angles usually result in constructing
a protein with a considerably different 3D structure
compared with its real protein. Here, to represent ac-
curate 3D structure of all atoms for an amino acid with
more than 20 atoms, 60 real variables –three coordi-
nates per atom– is needed. Therefore, if only value of
five torsion angles are used to reconstruct this protein,
a large amount of information must be presumed. In
this case, reconstructing not only involves the use of
protein’s torsion angles but also fairly accurate pre-
sumptions of its known bond lengths (mostly fixed)
and angles.
This work presents a method to minimize
the difference between the original and the re-
made/reconstructed protein by optimizing torsion an-
gles so that they absorb most noises in known angles
and lengths. Thus, the optimized torsion angles can
be used to extract useful information to facilitate fu-
ture PSP procedures. To present our work, section
2 describes our procedure, section 3 demonstrate our
experimental results followed by conclusions in sec-
tion 4.
2 PROCEDURE FOR TORSION
ANGLES REFINEMENT
Whenever torsion angles mathematically ob-
tained/calculated are deployed with known angles and
bond lengths, the differences between 3D structure
of the original protein and its remade/reconstructed
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
298
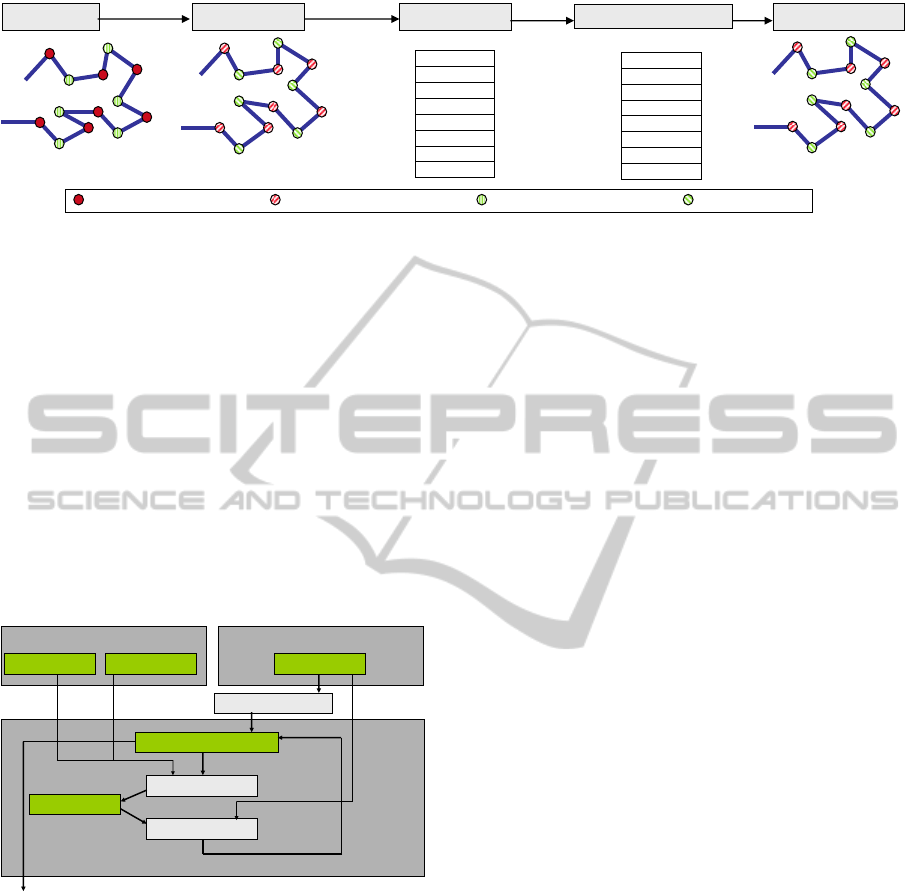
Noisy torsion angle Real static angle
Real torsion angle Noisy static angle
Real protein Noisy protein Remade protein
X-Ray
Torsion
Angles
extraction
PDB file
Torsion angles
3.24
2.45
56.13
-120.56
-75.25
170.10
17.90
…
Remaking
a) b) c) d)
Noisy torsion angle Real static angle
Real torsion angle Noisy static angle
Real protein Noisy protein Remade protein
X-Ray
Torsion
Angles
extraction
PDB file
Torsion angles
3.24
2.45
56.13
-120.56
-75.25
170.10
17.90
…
Optimization
Process
a) b) c) d)
Optimized Torsion angles
3.11
2.41
54.13
-121.22
-72.45
170.47
16.90
…
e)
Remaking
Figure 4: (a), (b) and (c) correspond to (a), (b) and (c) in Figure 3, (d) optimized torsion angles, (e) remade/reconstructed
protein using optimized torsion angles with more resemblance to the original protein.
one is considerably large. For instance, as shown in
Figure 3, a small amount of error in one part of the
protein can easily cause large errors in other parts.
This problem is even worsen –through increment of
cumulative noises– when most procedures that use
torsion angles assume the ideal value of 180 degrees
for their omega torsion angles. Figure 4 shows how
optimizing torsion angles to absorb the noise can
result in remaking/reconstructing a more similar
protein to its original PDB file compare with the
one remade using only the mathematically computed
torsion angles extracted from the data bank (Figure
3).
Protein Data BankBiology knowledge
Optimization process
PDB file
Mathematical method
Fixed angles Bond lengths
Torsion angles
Remade PDB file
Calculate Noise
New PDB file
Torsion angles optimization
Figure 5: Steps of our proposed procedure to optimize tor-
sion angles values.
Figure 5 presents the main steps of our proposed
procedure in this work to refine the torsion angles.
This procedure (1) uses the PDB file along with the
known bond lengths and fixes angles of the protein
structure as they are not heavily affected by noise sim-
ilar to the rest of torsion angles in a PDB file; and
then, (2) applies an optimization procedure (either a
gradient descent process or an evolutionary strategy)
to find the best set of torsion angles. It is also worth-
while mentioning that having torsion angles that can
fairly represent a protein plays an important role in
extracting its statistical information to summarize its
attributes. Therefore, if a protein cannot be fairly re-
made/reconstructed from its torsion angles, its statis-
tical analysis would also be based on noisy informa-
tion. and thus not very reliable.
Although 3D shape of a protein from a PDB file
could be significantly different from its remade ver-
sion given its torsion angles, their difference can be
overcome sometimes as it is is mainly causes by the
accumulative behavior of a large number of small er-
rors. Therefore, initial value of each torsion angle
variable must be fairly close to its optimal value, how-
ever must be further adjusted to absorb the noise.
Therefore, the best strategy to refine torsion angles
seems to be based on local searches. In this work, we
designed our algorithm based on two local search al-
gorithms: (1) the gradient descent algorithm to bench-
mark our results, and (2) the CMA-ES (Covariance
Matrix Adaptation Evolution Strategy) (Kern et al.,
2004; Hansen, 2006) as one of the best local search
algorithms reported to date.
3 RESULTS
Proteins 1PLW, 1CRN, 1UTG (RCSB, 2009) and
T0513 (CASP8) are used to gauge the performance
of our algorithm in this work. The protein 1PLW, also
known as enkephalin, has 5 amino acids with 22 tor-
sion angles, 1CRN has 46 amino acids with 194 tor-
sion angles, 1UTG has 72 amino acids with around
342 torsion angles, T0513 has 69 amino acids with
338 torsion angles, and T0496 has 120 amino acids
with 674 torsion angles to optimize. To obtain reli-
able results, each method is deployed for more than
20 times; we observed less than 3% deviation in their
results. Table 1 shows that our procedure managed to
reduce noises of 1CRN, 1UTG, and T0513 for more
than 70%, 80%, and 90%, respectively. Depending
on the time and the local search algorithm, different
torsion angles qualities were obtained.
Figure 6 shows a very challenging case of T0496
A METHOD TO IMPROVE THE ACCURACY OF PROTEIN TORSION ANGLES
299
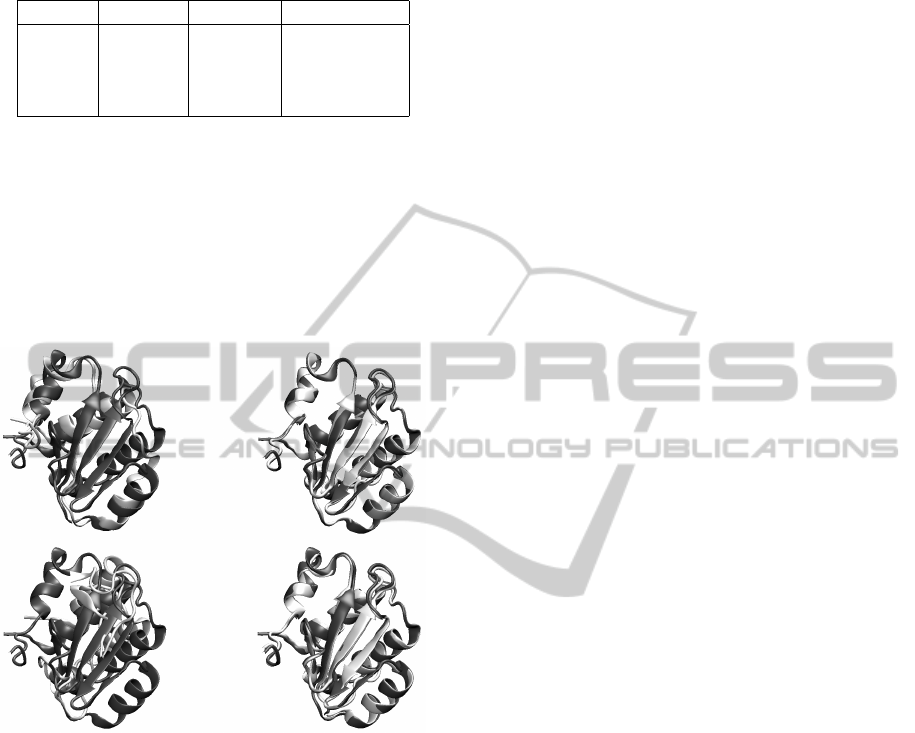
Table 1: RMSD between real protein and remade protein,
using original torsion angles and optimized torsion angles.
Protein Original CMAES Time (hours)
1PLW 0.908 0.789 0.23
1CRN 1.627 0.474 5.00
1UTG 4.862 0.610 6.70
T0513 7.215 0.715 6.56
with 120 amino acids from PDB where the cumula-
tive noise managed to result in a significantly different
protein structure –i.e., the remade proteins using the
original torsion angles result in a completely differ-
ent protein. Here, using our proposed algorithm, the
remade/reconstructed protein is much more similar to
the real protein: in both cases of considering and not
considering ideal omega angles.
Figure 6: Improvements in remaking the T0496 protein.
Each figure is a match of a real protein with a remade pro-
tein using: [left] mathematical torsion angles; [right] opti-
mized torsion angles obtained with CMA-ES; [top] consid-
ering omega torsion angles; [bottom] ignoring omega tor-
sion angle by setting them to their ideal values of 180 de-
grees.
In summary, results reveal that the less torsion
angles available, the less improvement could be
achieved by our procedure. This is mainly because,
in short proteins (with less than 20 amino acids) that
not many errors exists, remake/reconstructed proteins
could fairly resemble the overall structure of the orig-
inal protein; whereas, in large proteins (more than
100 amino acids) that accumulative errors are domi-
nant, small amounts of correction in one torsion an-
gle can significantly improve the quality of the re-
made/reconstructed protein to manifest better resem-
blance.
4 CONCLUSIONS
This work presents a framework to refine torsion an-
gles to remade/reconstruct more resemblant proteins
with similar 3D structures with their original proteins
from Protein Data Bank (PDB). This method deploys
local search algorithms such as gradient descent-
based approaches and/or evolutionary strategies to in-
corporate information in PDB to solve the infamous
Protein Structure Prediction (PSP) problem. Simula-
tion results of our algorithms showed that it can ef-
fectively reduce the accumulative noise behavior of
reconstructing proteins using their stored torsion an-
gles in PDB. Although reconstruction of small pro-
teins could also be improved (around 70%) using our
approach, the level of improvement in large proteins
was much more significant (more than 90%). This
framework can result in significantly more accurate
3D representation of proteins in PDB; and therefore,
can have very positive impacts in solving the PSP
problem.
ACKNOWLEDGEMENTS
This paper has been supported by the Spanish Minis-
terio de Educacion y Ciencia under project SAF2010-
20558.
REFERENCES
Hansen, N. (2006). The CMA evolution strategy: a compar-
ing review. In Lozano, J., Larranaga, P., Inza, I., and
Bengoetxea, E., editors, Towards a new evolutionary
computation. Advances on estimation of distribution
algorithms, pages 75–102. Springer.
Kern, S., M
¨
uller, S., Hansen, N., B
¨
uche, D., Ocenasek, J.,
and Koumoutsakos, P. (2004). Learning probability
distributions in continuous evolutionary algorithms–
a comparative review. Natural Computing, 3(1):77–
112.
Lesk, A. M. (2002). Introduction to Bioinformatics. Oxford
University Press. ISBN 0–19–927787-7.
RCSB (2009). Pdb (protein data bank).
UniProt, T. (2008). The universal protein resource (uniprot)
2009. Nucleic Acids Res., 37:169–174.
Zhang, Y. and Skolnick, J. (2005). The protein structure
prediction problem could be solved using the current
pdb library. Proc Natl Acad Sci USA, 102:1029–1034.
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
300
