
IMPROVED BREAST CANCER PROGNOSIS
BASED ON A HYBRID MARKER SELECTION APPROACH
L. Hedjazi, M.-V. Le Lann, T. Kempowsky-Hamon
CNRS, LAAS, 7, avenue du Colonel Roche, F-31077 Toulouse, France
Université de Toulouse, UPS, INSA, INP, ISAE, LAAS, F-31077 Toulouse, France
F. Dalenc, G. Favre
INSERM U563 and Institut Claudius Regaud, Toulouse, France
Keywords: Feature selection, Fuzzy logic, Mixed-Type Data, Breast cancer prognosis.
Abstract: Clinical factors, such as patient age and histo-pathological state, are still the basis of day-to-day decision for
cancer management. However, with the high throughput technology, gene expression profiling and
proteomic sequences have known recently a widespread use for cancer and other diseases management. We
aim through this work to assess the importance of using both types of data to improve the breast cancer
prognosis. Nevertheless, two challenges are faced for the integration of both types of information: high-
dimensionality and heterogeneity of data. The first challenge is due to the presence of a large amount of
irrelevant genes in microarray data whereas the second is related to the presence of mixed-type data
(quantitative, qualitative and interval) in the clinical data. In this paper, an efficient fuzzy feature selection
algorithm is used to alleviate simultaneously both challenges. The obtained results prove the effectiveness
of the proposed approach.
1 INTRODUCTION
Breast cancer is one of the most common causes of
death among women in the world. In 2009, an
estimation of 192,370 new cases of invasive breast
cancer was diagnosed, as well as 62,280 additional
cases of in situ breast cancer in the United States
alone. Along with 40,170 women are expected to die
from breast cancer and 1,910 cases of breast cancer
are expected to occur among men (data from the
American Cancer Society, 2009). Consequently, an
accurate cancer diagnosis and prognosis is needed to
help physicians take the necessary treatment
decisions and thereby reduce its related expensive
medical costs. In the past decade microarray analysis
has had a great interest in cancer management
(Golub et al., 1999; Ramaswamy et al., 2001; Van’t
Veer et al., 2002). This technology allowed a more
accurate cancer management such as diagnosis
(Ramaswamy et al., 2001), prognosis (Van’t Veer et
al., 2002), treatment response prediction (Straver et
al., 2009). Meanwhile, the introduction of this
technology has brought with it also new challenges
related to the high dimensionality of microarray data
and the low signal-to-noise ratio. During the pre-
microarray era, cancer management was guided by
the clinical and histo-pathological knowledge gained
from many decades of cancer research. It has been
established recently that the integration of both
information may improve the cancer management
(Sun et al., 2007; Gevaert et al., 2006). In (Sun,
2007), a feature selection method (I-Relief) was
used to perform markers selection. However, the
used method works under the assumption that all the
data are of quantitative type and therefore an
arbitrary transformation of symbolic data to
quantitative one was performed to cope with data
heterogeneity. This transformation can be a source
of distortion and information loss as it introduces a
distance which was not present in the original data.
In (Gevaert et al., 2006), a Bayesian network was
used to perform breast cancer prognosis. The
obtained results show only that their approach
performs similarly to the 70-gene signature
established by Van’t Veer and colleagues (Van’t
159
Hedjazi L., Le Lann M., Kempowsky-Hamon T., Dalenc F. and Favre G..
IMPROVED BREAST CANCER PROGNOSIS BASED ON A HYBRID MARKER SELECTION APPROACH.
DOI: 10.5220/0003152301590164
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2011), pages 159-164
ISBN: 978-989-8425-36-2
Copyright
c
2011 SCITEPRESS (Science and Technology Publications, Lda.)
Veer et al., 2002) and claim that a variable selection
is implicitly performed based on their (in)
dependency through the Markov Blanket concept.
These results do not mean necessarily that the
clinical data contains no additional information to
the genetic data; it only tells us that their approach
does not fit well. In the present work, we use our
recently developed method, referred to as MEMBAS
for (MEmbership Margine Based FeAture Selection)
(Hedjazi et al., 2010a), to prove the usefulness of the
integration of both types of data by handling both
challenges simultaneously: high-dimensionality and
heterogeneity of data. The first challenge is one of
the characteristic of microarray data related to the
curse of dimensionality (Golub et al., 1999). To deal
with this problem, MEMBAS method selects a small
feature subset such that the performance of a
learning algorithm is optimized. The second
challenge concerns the problem of processing
simultaneously different types of data (qualitative,
quantitative, interval ...) present almost in all daily
produced clinical datasets (Age, Sex, Tumour size,
Tumour grade...). MEMBAS method answers also to
this problem by a simultaneous mapping of all types
of data on a homogeneous space in order to process
them identically in this new resulted space.
This paper is organized as follows: the second
section explains the fuzzy feature selection approach
based on feature fuzzification and the MEMBAS
selection procedure. An application is given in
section 3 to prove the usefulness of the adopted
approach through the derivation of a hybrid
signature for breast cancer prognosis.
2 FUZZY FEATURE SELECTION
During the past decades, feature selection has played
a crucial role in order to improve the learning
algorithms performance by selecting only the most
relevant features for the problem under
investigation. Here, we use the term feature to refer
to a marker. Existing feature selection algorithms are
traditionally characterized as wrappers and filters
according to the criterion used to search the relevant
features (Kohavi and John, 1997; Guyon and
Elisseeff, 2003). Wrapper algorithms optimize the
performance of a specified machine-learning
algorithm to assess the usefulness of the selected
feature subset; whereas filter algorithms use an
independent evaluation function based generally on
a measure of information content (entropy, t-test,…)
(Kohavi and John, 1997; Guyon and Elisseeff,
2003). Filter algorithms are computationally more
efficient but perform worse than wrapper algorithms
(Kohavi and John, 1997; Guyon and Elisseeff,
2003). Thereby, with filter algorithms the features
are evaluated individually without taking into
account the correlation information and redundancy
problems. Hence, this can deteriorate drastically the
classifier performance (Kohavi and John, 1997). On
the other hand, daily produced medical datasets may
contain mixed feature-types (numerical, symbolic
data) as well as large number of irrelevant features.
This also poses a great challenge for the existing
machine-learning algorithms. Up to now, most
classical feature selection algorithms are suitable for
numerical features but their efficiency decreases
significantly whenever a mixed-type dataset problem
is encountered. The second problem is assessed in
emergent fields such as bioinformatics, where
datasets may hold a huge number of irrelevant
features.
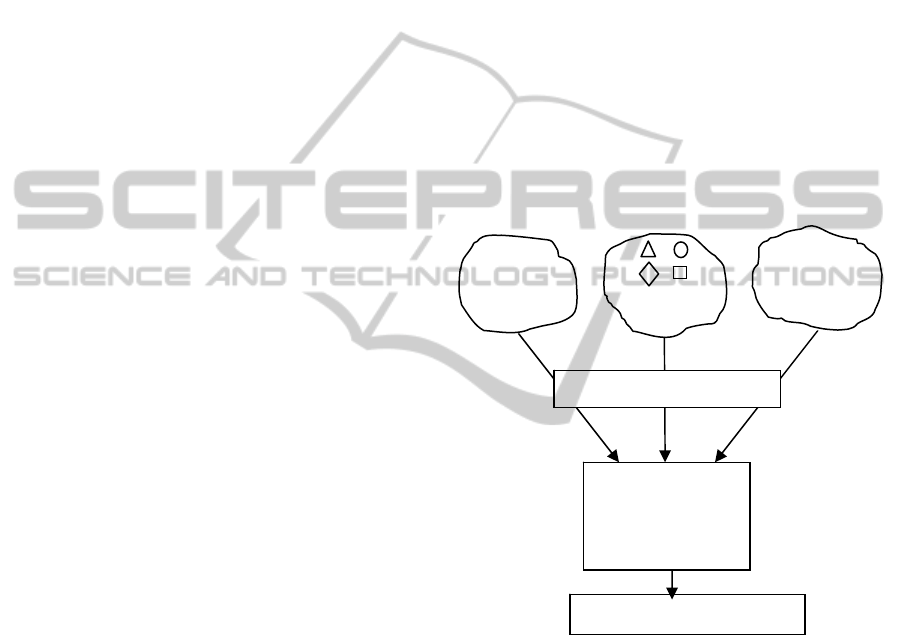
Figure 1: MEMBAS general principle.
We have recently proposed a new feature
selection algorithm, referred to as MEMBAS
(Hedjazi et al., 2010a), which alleviates the
previously mentioned problems. MEMBAS enables
to process in the same way the three types of data
(numerical, qualitative and interval) based on an
appropriate and simultaneous mapping using fuzzy
logic concepts (Figure 1.). To avoid the heuristic
search during the feature selection procedure,
MEMBAS optimizes an objective function using
classical optimization techniques. The feature’s
importance is therefore evaluated within a
membership margin framework. As we address a
problem with two classes (Recurrence or No
1.0 0.25 3.0
0.5 2.5 0.9
Qualitative
Space
Interval Space
[0.35 1] [1.2 4]
[1.45 3] [1.4 2]
[µ
11
µ
12
… … µ
1m
]
[µ
21
µ
22
… …
µ
2m
]
… … … … … …
Membership
S
Simultaneous mapping
Single processing
Quantitative
Space
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
160

Recurrence), only a description of MEMBAS for
binary class problems is given in this paper.
Let
CC
n
×Χ∈
=
N
1nn
],[x =D
be the training dataset,
where x
n
= [x
n1
, x
n2
, ..., x
nm
] is the n-th data sample
containing m features, and C
n
its corresponding class
label. In the first step the features subset is fuzzified
using the empirical data based on the appropriate
learning process according to each feature type. The
resulting fuzzy sets represent the feature
memberships to the two existing classes. Then, when
a new representation of data in a homogeneous
space “membership space” is obtained, a single
processing can be performed whatever the initial
type of data.
2.1 Feature Fuzzification
The feature fuzzification can be performed
according to each feature type as follows:
2.1.1 Quantitative Type Features
The quantitative feature value is first normalized
into the interval [0,1] by using the formula:
min max min
ˆˆ ˆ ˆ
/
ii i i
xxx x x
i
=− −
(1)
Where
ˆ
i
x
is the measured value of the i
th
feature
and
i
x
is its normalized value, x
imin
and x
imax
are the
bounds of the i
th
feature given by the context or
imposed by the expert.
In the case of quantitative features, several
membership functions proposed by (Aguado and
Aguilar, 1999)
can be used for µ
k
i
. In this work we
use the centred binomial membership function (2):
[]
()
i
ki
i
ki
x
i
k
x
i
k
i
k
i
ki
i
k
x
ρρ
ϕϕϕρμ
−−−
−= 1,
1
(2)
where
k
ρ
is the i
th
feature prototype for class C
k
,
and parameter
k
ϕ
measures the proximity of the
feature value to the class prototype so that :
⎥
⎦
⎤
⎢
⎣
⎡
≥
⎥
⎦
⎤
⎢
⎣
⎡
≠∀
i
k
i
ki
i
k
i
k
i
k
i
k
i
k
i
ki
xx
ϕρμϕρρμρ
,,:
and for
i
k
i
x
ρϕϕ
≠∀≤
21
, we have the ordered
memberships
⎥
⎦
⎤
⎢
⎣
⎡
≥
⎥
⎦
⎤
⎢
⎣
⎡
12
,,
ϕρμϕρμ
i
ki
i
k
i
ki
i
k
xx
.
2.1.2 Interval Type Features
The membership function for interval type variables
is chosen as the similarity (Hedjazi et al., 2010b)
between the symbolic interval value of the i
th
variable x
i
and the interval
,
i
k
ii
kk
ρρρ
−+
=
⎡⎤
⎣⎦
representing
the class C
k
as:
(
)
(
)
i
k
i
i
i
k
x
S
x
ρμ
,=
(3)
Given 2 intervals
[
]
−+
= aaA , and
[
]
−+
= bbB , ,
their distance
∂
is defined as:
[]
{
}
{
}
(
)
,,,max0,max minab abAB
−− ++
∂= −
⎡
⎤
⎣
⎦
The similarity measure between two intervals A and
B is defined as:
()
[
]
[]
[]
[]
⎟
⎠
⎞
⎜
⎝
⎛
∂
−+
∪
∩
=
U
BA
BA
BA
BAS
ϖϖ
ϖ
,
1
2
1
,
(4)
Where the measure
ϖ
of an interval
X
is given by:
[
]
(
)
(
)
XboundlowerXboundupperX .. −=
ϖ
Let consider that m
k
individuals have been assigned
to class C
k
, this class will have as prototype a vector
whose components are the intervals obtained by the
mean bounds:
∑
=
−−
=
k
m
j
j
i
k
i
k
x
m
1
)(
1
ρ
and
∑
=
++
=
k
m
j
j
i
k
i
k
x
m
1
)(
1
ρ
(5)
Where
j
i
x
−
is the i
th
variable lower bound of the j
th
sample and
j
i
x
+
is its upper bound. Consequently,
the resulted class prototype for the r interval
variables is given by the vector of intervals:
[
]
T
r
kkk
k
ρρρρ
,...,,
21
=
(6)
For a better conditioning of magnitudes and
processing time minimization, normalization within
the interval [0,1] is proposed:
−+
−+
+
−+
−−
−
−
−
=
−
−
=
minmax
min
minmax
min
ˆˆ
ˆˆ
,
ˆˆ
ˆˆ
ii
ii
i
ii
ii
i
xx
xx
x
xx
xx
x
(7)
where
],[x
+−
=
iii
xx
is the normalized value;
consequently, the domain U
i
of any interval variable
x
i
becomes the unit interval [0,1].
2.1.3 Qualitative Type Features
For qualitative variables, the possible values of the
i
th
variable form a set of modalities:
IMPROVED BREAST CANCER PROGNOSIS BASED ON A HYBRID MARKER SELECTION APPROACH
161

{
}
1
,,
iii
ijMi
DQQQ=……
(8)
The membership function for a qualitative variable x
i
is specified as:
(
)
(
)
(
)
1
1
iiMi
qq
ii i
k k kMi
i
x
μ
=Φ ∗ ∗Φ
(9)
Where
i
kj
Φ
is the frequency of modality
i
j
Q
in the
class C
k
and
⎩
⎨
⎧
≠
=
=
i
ji
i
ji
i
j
Qx if
Qx if
q
0
1
. Therefore, the class
prototypes are represented by
1
,, ,,
ii i i
kk kj kMi
⎡⎤
Ω=Φ Φ Φ
⎣⎦
……
.
2.2 Homogeneous Space of Features
It results from the previous step that, in the binary
class problems, a sample x
n
from dataset D can be
associated to two Membership Degree Vectors
(MDVs) of dimension
m given as follows:
() () ( )
[]
1,2.= ;
T
x ,...,x ,xU
nm
m
n2
2
n1
1
nc
k
kkk
k
μμμ
=
(10)
Where
µ
k
i
(x
ni
) (i.e. µ
k
i
(x
i
= x
ni
) ), is the membership
function of class C
k
evaluated at the given value x
ni
of the i
th
feature for sample x
n
.
It is worthwhile to recall that the resulted MDVs
contain the membership values relative to all
features whatever their initial type. This guaranties
the mapping of different feature types from
completely heterogeneous spaces into a common
space which is the membership space (Figure 1).
Once all features are simultaneously mapped into a
common space, they can be henceforth processed
similarly either for a classification or feature
selection task. Our focus in this work is the feature
selection task. A membership margin has been
introduced in (Hedjazi
et al., 2010a) to estimate the
features importance in the membership space
whatever their type and number. We assume that the
n
th
data sample
12
n
x,,
m
nn n
xx x=
⎡⎤
⎣⎦
is labelled
by class
c
. Let c
~
be the alternative class. We define
a membership margin for sample x
n
by:
(
)
(
)
n
β = ψ U-ψ U
nc nc
(11)
where
U
nc
and
U
nc
are respectively the
membership degree vectors of sample x
n
to classes
c
and c
~
, ψ(.) is an aggregation function defined as
∑
=Ψ
i
i
Y (Y)
computing the global contribution of a
subset of features to each class. Note that a sample
x
n
is correctly classified if β
n
>0. The basic idea to
calculate the fuzzy feature weight is to scale the
feature memberships in the membership space such
that the leave-one-out error is minimized:
(
)
(
)
Max β (w ) { }
1n f 1 1 1
w
f
NNmimi
wx wx
n n i fic ni i fic ni
μμ
∑=∑∑ −∑
=== =
0 w1,=|| w|| t S.
f
2
2f
≥
(12)
Where β
n
(w
f
) is the margin of x
n
computed with
respect to w
f
. The first constraint is the normalized
bound for the modulus of w
f
so that the
maximization ends up with non infinite values,
whereas the second guarantees the nonnegative
property of the obtained weight vector. The classical
Lagrangian optimization approach was used to solve
the above problem and the following closed-form
solution was obtained:
*
f
s
w
|| s ||
+
+
=
{}
∑
=
−=
N
n
cnnc
1
~
U U s where
(13)
With s
+
= [max(s
1
,0), …, max(s
m
,0)]
T
.
Therefore, MEMBAS is considered as one of the
first feature selection algorithms that enable
processing similarly mixed feature-type data. In
addition, the objective function optimized by
MEMBAS approximates the leave-one-out cross
validation error. Therefore, MEMBAS chooses only
the features if they contribute to the overall
performance. Hence, it addresses the issues of
features correlation and redundancy. Moreover,
MEMBAS avoids the heuristic combinatorial search
by using classical optimization approaches to
achieve an analytical solution. In (Hedjazi
et al.,
2010a) an extensive experimental study was
performed on large number of datasets presenting
both challenges (mixed-type and high-dimensional
data) to demonstrate the effectiveness of the
algorithm. The novelty of the present study is the
application of this method to derive a hybrid
signature integrating simultaneously genes
expression and heterogeneous clinical data
(quantitative, qualitative, interval). Moreover, an
extension of MEMBAS method has been also
proposed for multiclass problems (Hedjazi
et al.,
2010a). Subsequently, the effectiveness of
MEMBAS method is illustrated on a real-world
problem of crucial importance: marker selection for
breast cancer prognosis. The main aim for
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
162

performing this study is to improve cancer prognosis
based on the dimensionality reduction principle
when the data are possibly of mixed types. As it was
mentioned in the previous section, MEMBAS
enables a simultaneous selection of mixed-feature
types by avoiding any related numerical and
heuristic search complexities.
3 EXPERIMENTS AND RESULTS
3.1 Dataset and Experiment Setup
The data set used in this study consists of 295 breast
cancer patients, divided into 2 classes according to
the appearance of distant subclinical metastases: 88
patients with and 207 patients without distant
metastases (Van de Vijver et al., 2002). 29 patients
with missing data have been removed. The
microarray data set contained 24188 gene expression
values. The clinical data contained 11 variables:
•
Age (quantitative)
•
Tumour grade (interval: [3,5]; [6,7]; [8,9])
•
Tumour size = T (qualitative: ≤2cm; >2cm)
•
Nodal status = N (qualitative : pN0; ‘1-3’; ≥4)
•
Mastectomy (qualitative : Yes, No)
•
Estrogen Receptor expression (qualitative: Yes,
No)
•
Chemotherapy (qualitative: Yes, No)
•
Hormonotherapy (qualitative: Yes, No)
•
St. Gallen - European criteria (qualitative:
Chemo, No Chemo)
•
NIH –US criteria (qualitative: Chemo, No
Chemo
•
Risk NIH (qualitative: low, intermediate, high)
The complete data set (clinical and microarray data)
was divided, similarly as in (Chang
et al., 2005),
into a training set (132 patients) to perform feature
selection and learn classifier parameters, and a
validation set (134 patients) to assess the
performance of the algorithm on data not used for
training. The classification task was performed by
using the fuzzy classification algorithm LAMDA
(Learning Algorithm of Multivariate Data Analysis)
(Aguado and Aguilar-Martin, 1999). LAMDA is a
fuzzy methodology of conceptual clustering and
classification. It is based on finding the global
membership degree of a sample to an existing class,
considering all the contributions of each of its
features. This contribution is called the
marginal
adequacy degree
(MAD). The MADs are combined
using "fuzzy mixed connectives" as aggregation
operators in order to obtain the
global adequacy
degree
(GAD) of an element to a class. We have
chosen this classifier because it handles in a unified
way the three types of data (quantitative, qualitative
and interval) without the need of any transformation.
More details on this fuzzy classification method can
be found in (Hedjazi
et al., 2010b) which did not
address the feature selection problem. MEMBAS is
used here to derive a hybrid prognostic marker
without resorting to any data transformation. To
demonstrate the predictive power of the hybrid
prognostic signature derived from the genetic and
clinical markers, its performance was compared with
those of clinical markers and the well known
Amsterdam 70-genes signature (Van’t Veer, 2002).
Then, another comparison with purely clinical
indices (NIH, St Gallen) was also performed.
3.2 Results
Table 1 shows the obtained comparative results
between the hybrid markers approach and other
approaches. It can be observed that the best
prediction accuracy is obtained by the proposed
approach which achieves more than 70%, whereas
only 66% is achieved using the 70-genes
Amsterdam signature (Van’t Veer, 2002). It must be
noticed here that MEMBAS chooses only 15 hybrid
markers, among them three are mixed-type clinical
markers (Number of positive lymph nodes
“qualitative” , Estrogen Receptor “qualitative” and
Grade “interval”), added to them 12 genes. This fact
was established in many previous studies (Deepa
and Claudine, 2005), where it was noted that these
three clinical features still to date are considered as
important prognostic factors. Therefore, MEMBAS
chooses meaningful markers and allows reducing
significantly the number of needed markers to
perform a prognosis (12 genes compared to the 70
genes of the Amsterdam signature).
Table 1: Comparatives results between hybrid, clinical and
genetic signatures.
TP FP FN TN Sens Spec Acc
Hybrid 13 12 28 81 0.32 0.87
94/134
(70.15%)
70-genes 25 29 16 64 0.61 0.69
89/134
(66.42%)
Clinical 23 37 18 56 0.56 0.60
79/134
(58.96%)
To further demonstrate the effectiveness of the
proposed approach, we compare in Table 2 our
results with the following clinical conventional
prognostic factors: the St. Gallen’s European
consensus and the NIH index. The St. Gallen and the
IMPROVED BREAST CANCER PROGNOSIS BASED ON A HYBRID MARKER SELECTION APPROACH
163

NIH prognostics were taken from the clinical dataset
as given by (Chang
et al., 2005).
Table 2: Comparative results between hybrid markers and
pure clinical indices (NIH, St Gallen).
TP FP FN TN Sens Spec Acc
Hybrid 13 12 28 81 0.32 0.87
94/134
(70.15%)
NIH 41 91 0 2 1 0.02
41/134
(32.09%)
St
Gallen
38 85 3 8 0.93 0.09
46/134
(34.33%)
Both indices have a very high sensitivity, but an
intolerable low specificity which would lead to give
unnecessary adjuvant systematic treatment to many
patients. Thus the obtained hybrid markers
outperforms also the pure clinically indices.
4 CONCLUSIONS
In this paper a new approach to perform cancer
prognosis is proposed based on a hybrid marker
selection. We evaluated our approach on a public
available breast cancer prognosis dataset. Patients
included in this dataset are classified into two groups
according to whether a distant subclinical metastasis
was occurred or not. This dataset represents two
challenges: high-dimensionality (microarray data)
and mixed-type data (clinical data). To cope
appropriately with this, a marker selection was
performed based on a fuzzy feature selection
approach which handles both challenges. It has been
shown that the obtained hybrid markers, composed
of clinical markers and genes, can improve the
prediction accuracy and outperform both genetic
based approaches (i.e. the well-known Amsterdam
70-genes signature) and pure clinical indices (St
Gallen and NIH). Moreover, the proposed approach
reduces significantly the number of markers needed
to perform a cancer prognosis task.
Future work will be devoted to test this algorithm on
other public available datasets and integrate other
sources of information than clinical and microarray
data.
REFERENCES
Aguado J. C., and Aguilar-Martin J., 1999. A mixed
qualitative-quantitative self-learning classification
technique applied to diagnosis, QR'99 The Thirteenth
International Workshop on Qualitative Reasoning.
Chris Price, 124-128.
Chang H. Y., Nuyten D. S. A., Sneddon J. B., Hastie T.,
Tibshirani R., et al., 2005. Robustness, scalability, and
integration of a wound-response gene expression
signature in predicting breast cancer survival. PNAS
2005, 102 (10): 3738-3743
Deepa, S., Claudine I., 2005. Utilizing Prognostic and
Predictive Factors in Breast Cancer. Current
Treatment Options in Oncology 2005, 6:147-159.
Current Science Inc
Gevaert O., De Smet F., Timmerman D., Moreau Y., De
Moor B., 2006. Predicting the prognosis of breast
cancer by integrating clinical and microarray data with
Bayesian network, Bioinformatics 22 (14), 184-190.
Golub T. , Slonim D., Tamayo P., Huard C., Gaasenbeek
M., et al., 1999. Molecular Classification of Cancer
Class Discovery and Class Prediction by Gene
Expression Monitoring, Science 286 (5439), 531-537.
Guyon I., Elisseeff A., 2003. An introduction to variable
and feature selection, J. Mach. Learn. Res, 3, 1157-
1182.
Hedjazi L., Aguilar-Martin J., Le Lann M. V., and
Kempowsky T., 2010a, Membership-Margin based
Feature Selection for Mixed-Type and High-
Dimensional Data, Fuzzy Sets and Systems Journal.,
submitted for publication.
Hedjazi, L., Kempowsky T., Le Lann M. V., Aguilar-
Martin J., 2010b. Prognosis of breast cancer based on
a fuzzy classification method. 3rd International Joint
Conference on Biomedical Engineering Systems and
Technologies (BIOSTEC 2010); 1st International
Conference on Bioinformatics (BIOINFORMATICS
2010). Valence (Spain), 20-23 January 2010, pp.123-
130.
Kohavi R., and John G. H., 1997. Wrapper for feature
subset selection, Artificial Intelligence 97, 273-324.
Ramaswamy S., Tamayo P., Rifkin R., Mukherjee S.,
Yeang C., et al., 2001. MultiClass Cancer Diagnosis
Using Tumor Gene Expression Signatures, Proc. Nat’l
Acad. Sc. USA 98 ( 26) , 15149-15154.
Straver M. E., Glas A. M., Hannemann J., Wesseling J.,
van de Vijver M. J., et al.,2009. The 70-gene signature
as a response predictor for neoadjuvant chemotherapy
in breast cancer, Breast Cancer Res. Treat.,
doi:10.1007/s10549-009-0333-
Sun Y., 2007. Iterative RELIEF for Feature Weighting:
Algorithms, Theories, and Applications, IEEE TPAMI,
2 (6) , 1035-1051.
Sun Y., Goodison S., Li J., Liu L., Farmerie W., 2007.
Improved breast cancer prognosis through the
combination of clinical and genetic markers.
Bioinformatics, Gene expression. Oxford University
Press 23 (1), 30-37.
Van’t Veer L.J., Dai H., van de Vijver M. J., et al., 2002.
Gene expression profiling predicts clinical outcome of
breast cancer, Nature, 415, pp. 530-536.
Van de Vijver M. J., He Y. D., Van’t Veer L. J., Dai H.,
Hart A., et al., 2002. A Gene expression signature as a
predictor of survival in breast cancer, N Engl J Med,
347 (25), pp. 1999-2009.
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
164
