
KINETIC MODELS AND QUALITATIVE ABSTRACTION
FOR RELATIONAL LEARNING IN SYSTEMS BIOLOGY
Gabriel Synnaeve
1
, Katsumi Inoue
2
, Andrei Doncescu
3
, Hidetomo Nabeshima
4
1
E-Motion Team at INRIA, Grenoble, France
2
National Institute of Informatics, Tokyo, Japan
3
LAAS-CNRS 31007, Toulouse, France
4
University of Yamanashi, Yamanashi, Japan
Yoshitaka Kameya, Masakazu Ishihata, Taisuke Sato
Tokyo Institute of Technology, Tokyo, Japan
Keywords:
Systems biology, Discretization, Metabolic pathways, Inductive logic programming, Abduction.
Abstract:
This paper presents a method for enabling the relational learning or inductive logic programming (ILP) frame-
work to deal with quantitative information from experimental data in systems biology. The study of systems
biology through ILP aims at improving the understanding of the physiological state of the cell and the interpre-
tation of the interactions between metabolites and signaling networks. A logical model of the glycolysis and
pentose phosphate pathways of E. Coli is proposed to support our method description. We explain our original
approach to building a symbolic model applied to kinetics based on Michaelis-Menten equation, starting with
the discretization of the changes in concentration of some of the metabolites over time into relevant levels. We
can then use them in our ILP-based model. Logical formulae on concentrations of some metabolites, which
could not be measured during the dynamic state, are produced through logical abduction. Finally, as this re-
sults in a large number of hypotheses, they are ranked with an expectation maximization algorithm working
on binary decision diagrams.
1 INTRODUCTION
Nowadays, systems biology represents the key field
to explain the functionality of life science. To ana-
lyze a biological system it is necessary to find out new
mathematical models allowing to explain the evolu-
tion of the system in a dynamic context or to deal in a
simple manner with the complex situations where the
human experience overtakes mathematical reasoning
(Kitano, 2002). Many physical and biological phe-
nomena may be represented on an analytical form us-
ing dynamical system. Our case study is based on
wet biology experiment consisting in applying a pulse
of glucose in a small bio-reactor containing E.Coli
that led to building an ordinary differential equations
(ODEs) based simulator. We used high performance
liquid chromatography to measure some metabolites
concentrations and some others had to be estimated,
using a simulated annealing algorithm, since no ex-
perimental results were available. So, knowing com-
pletely the evolutions of metabolites concentrations of
this system, we applied our approach to show its cor-
rectness. For that, we took only steady-state values of
metabolites concentrations and ran our model.
Several attemps have been done for logic-based
approaches to analyze biochemical pathways in Sys-
tems Biology. They use action languages (Baral et al.,
2004), abduction (Juvan et al., 2005; King et al.,
2004; King et al., 2005; Tamaddoni-Nezhad et al.,
2006), SAT (Tiwari et al., 2007), inductive logic pro-
gramming (Doncescu et al., 2007) or answer set pro-
gramming (Dworschak et al., 2008). All these pre-
vious approaches are based on qualitative modeling,
and none of them can handle continuous domains ap-
propriately. Temporal logic combined with the rep-
resentation of kinetic models in stochastic logic pro-
gramming (SLP) (Fages et al., 2008) have a simi-
lar goal using different means: the authors modeled
the kinetics of biochemical systems by continuous
time Markov chains as input to SLP where we took
an approach to discretize (through continuous HMM)
concentrations of metabolites first and then use them
combined with a logical translation of ODEs-based
kinetics as input to ILP. The goal of this research is
47
Synnaeve G., Inoue K., Doncescu A., Nabeshima H., Kameya Y., Ishihata M. and Sato T..
KINETIC MODELS AND QUALITATIVE ABSTRACTION FOR RELATIONAL LEARNING IN SYSTEMS BIOLOGY.
DOI: 10.5220/0003166300470054
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2011), pages 47-54
ISBN: 978-989-8425-36-2
Copyright
c
2011 SCITEPRESS (Science and Technology Publications, Lda.)
to incorporate continuous values and kinetics within
the logic-based approach to metabolic pathways. In
particular, we enhance an abductive framework pro-
posed in (Inoue et al., 2009), which consists of ab-
ductive hypothesis generation and statistical hypoth-
esis evaluation, by enabling us to handle real-valued
data obtained from measurement in observations.
For that, we now propose a loop for learning
about a metabolic pathway from experiments in which
we have to (each step corresponds to a section, as in
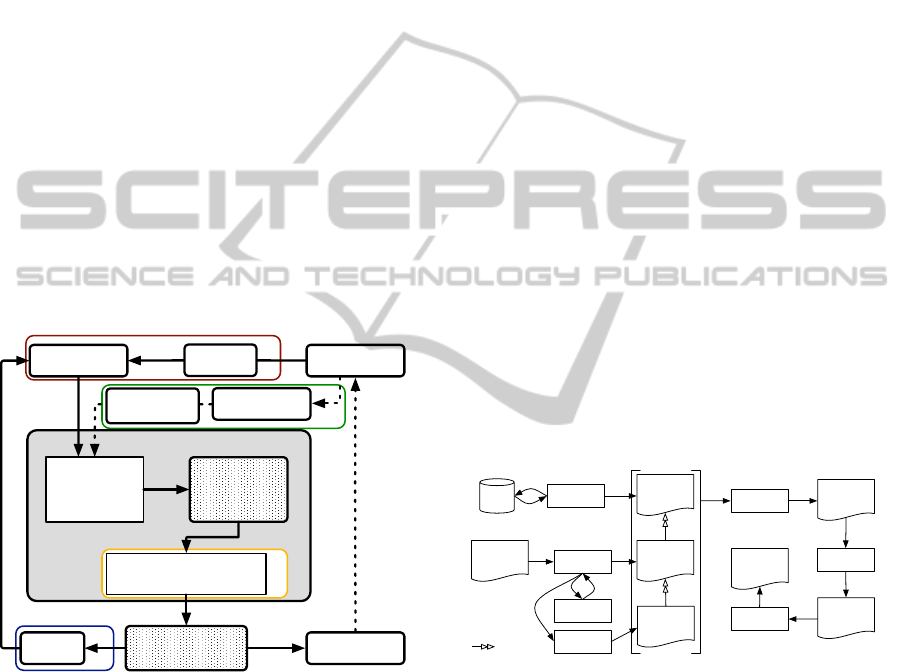
Fig. 1):
1. clusterize continuous concentrations of metabo-
lites over time into discrete levels and discrete
timesteps.
2. use them in an ILP-based model of the pathway,
in conjunction with a set of knowledge-generating
rules, here in the example describing Michaelis-
Menten kinetics.
3. sort the resulting abduced facts or inducted rules
with our defined metrics.
4. use this ranking for enhancing our knowledge
base and goto the beginning of this process.
4
Experiments
Logically
possible
hypotheses
Databases
Hypotheses
Generator
(SOLAR)
Hypotheses Evaluator
(BDD-EM)
Background
knowledge
Observations
Most probable
hypotheses
3
Discretization
Automated
Enhancer
1
2
Figure 1: Overview of the complete process.
In this paper, we show how this “closed loop”
architecture can be applied to an inverse problem:
given the measured concentrations of some metabo-
lites in a steady state, we compute the concentra-
tions of metabolites before the dynamic transition to
this steady state based on the kinetic modeling. We
worked with the beginning of an automated frame-
work (see Fig. 2 for a practical data-centric circuit)
to deal with different real world pathways and exper-
iments. It is mainly composed of four tools:
• The combination of an implementation of contin-
uous HMMs (Gauvain and Lee, 1994; Ji et al.,
2006) with PY-TSDISC to discretize experimental
values.
• KEGG2SYMB, using the KEGG API, that trans-
form pathways from KEGG (Kanehisa and Goto,
2000; Kanehisa et al., 2008) into symbolic mod-
els.
• SOLAR, a consequence finding system working
on Skipping Ordered Linear tableaux (Nabeshima
et al., 2003), which is complete for finding mini-
mal explanations, to conduct abduction or induc-
tion.
• BDD-EM, an implementation of the expectation-
maximization algorithm on binary decision dia-
grams (Ishihata et al., 2008; Inoue et al., 2009)
to rank hypotheses.
We chose to illustrate this method on the conjunc-
tion of glycolysis and pentose phosphate pathways for
E.Coli, simplified the model by keeping 16 relevant
reactions and discretized experimental values (16 val-
ues) as in section 1. We added the three Michaelis-
Menten based rules and the three constraints of unic-
ity for the levels as in section 2. We had 15 un-
known levels of concentrations of metabolites before
the transition to the steady state (yielding 15×3 levels
= 45 abducibles). SOLAR, used for abduction, outputs
98 hypotheses that cover all these metabolites. With
such a number, picking the right hypotheses should
be done in an automated way as we did in section 3.
KEGG
Experimental
Data
kegg2symb
Symbolic
pathway
py-tsdisc
HUP
SOLAR
Discretized
data
Kinetic rules
BDD-EM
Hypotheses
Ranked
hypotheses
: append
enhancer
New KB
rules
generator
Figure 2: Data-centric schema of the process.
2 DISCRETIZATION OF TIME
SERIES FROM EXPERIMENTS
In our modeling, we first introduce discrete concen-
tration levels to filter what are the relevant changes
of concentration of the metabolites, in regard to hy-
potheses generation from ILP. We need to be able to
infer hypotheses that have a certain level of generality
and, for that, we should use intervals instead of single
real values. This could have been done with an inter-
val constraints approach (Benhamou, 1994), but we
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
48

currently choose a discretization approach. Although
this gives us less freedom in the logic part as levels
are fixed (as if we have fixed intervals), levels can be
handled just as symbols in a logical model of path-
ways.
Discretizing time series is a research field in which
many works (Geurts, 2001; Keogh et al., 2005) have
been conducted recently. Our practical problem is that
we want to have a statistically relevant (unsupervised)
discretization for N metabolites concentrations over
time. We also discretize the values of K
m
(Michaelis-
Menten constants, see (1)), for each reaction, with the
same levels. For that purpose, we use a probabilis-
tic model, used in speech recognition and time series
analysis: continuous hidden Markov model (HMMs)
(Rabiner, 1989). We can therefore compute an ap-
propriate number of levels (that was three for E.Coli)
in regard to a Bayesian score such as Bayesian In-
formation Criterion (BIC) (Schwarz, 1978) or as the
Cheeseman-Stutz score (Cheeseman and Stutz, 1995)
or as the variational free energy. This process can
be achieved through the following methods all de-
scribed in (Beal, 2003), respectively: maximum like-
lihood estimation or maximum a posteriori estimation
or through a variational Bayesian method.
We use continuous (Gaussian) HMMs with pa-
rameter tying
1
. This is a solution to the problem of
sharing the same symbolic levels in all the logic mod-
els in order to be able to assign the level of a com-
pound to another and be dealing with the same real
values behind the scene. We first prepare N contin-
uous HMMs (one for each metabolite), where each
state variable takes a concentration level, and each
output variable takes a measurement of concentration
and follows a univariate Gaussian distribution. All the
HMMs share a state space as well as the parameters
in the output variables (i.e. means and variances), so
that they produce discrete levels that are correspond-
ing. These relevant discretized levels of concentration
are computed through the expectation-maximisation
(EM) algorithm with maximum a posteriori (MAP)
estimation (Gauvain and Lee, 1994) or through the
variational Bayes EM (VB-EM) (Beal, 2003; Ji et al.,
2006). We prefer this last method as it is shown (Beal,
2003) that variational free energy provides a more ac-
curate approximation of the marginal log-likelihood
than BIC or the Cheeseman-Stutz score.
1
Parameter tying is a notion often used in HMMs for
speech recognition (Rabiner, 1989) and recently in statisti-
cal relational learning (De Raedt, 2008). In our case, the
mean and the variance for X
(n)
t
, the output variable at time
t in the HMM for the n-th metabolite (n = 1,..., N), are
tied with the mean and the variance for X
(n
0
)
t
0
, respectively
(n 6= n
0
and t 6= t
0
).
Figure 3: 3-State continuous HMM discretizing one experi-
mental time series, where X
t
is the measurement of concen-
tration at time t and S
t
is the hidden state that indicates the
corresponding discretized level.
Then, we use a simple round-mean aggregation of
them for time-sampling. We set a maximal number
of time steps and look for the better fitting width and
alignment for equal-width time intervals. We are cur-
rently developping a different process in the direction
of discretization of our time series from molecular bi-
ology experiments that will discretize time and levels
simultaneously but current results are already useable
(see Table 1 and Fig. 5) and that is what we based the
work presented here on.
3 MODELING OF THE
PATHWAYS OF E.COLI
To obtain an understanding of the central metabolism,
a logical model has been developed according to a
kinetic model including the glycolysis and the pen-
tose phosphate pathway for Escherichia coli (Chas-
sagnole et al., 2006). The Fig.4 shows the simplified
pathway that we modelized logically with relations
reaction(Substrate, Enzyme, Product, Km).
pg6 ribu5p rib5p
glucose g6p xyl5p sed7p
f6p
dhap fdp pep pyr
gap pg3 accoa
cell membrane
Figure 4: Simplified glycolysis and pentose phosphate path-
ways for E.Coli.
The metabolic networks dynamics are in their en-
zymatic part ruled by the combination of classical
kinetics: essentially Michaelis-Menten, Hill and al-
losteric ones. If we limit our modeling to these kinet-
KINETIC MODELS AND QUALITATIVE ABSTRACTION FOR RELATIONAL LEARNING IN SYSTEMS
BIOLOGY
49

ics, we can highly simplify their mathematical han-
dling, and that is what we did. We chose to use only
Michaelis-Menten kinetics, because we had a path-
way simple enough and that it is the more general rep-
resentation for a non-linear allosteric regulation sys-
tem. It assumes that the two enzyme binding equi-
libria are fast when compared to the interconversion
of enzyme + substrate (ES) and enzyme + product
(EP) compounds. That assumption appears reason-
able considering that the dynamics of the experiment
were happening in less that a minute: this implies that
the effects of genetic regulation of the enzymes in-
cluded are negligible and so the maximum reaction
rates represent the amount and catalytic activity of en-
zymes.
E + S
k
1
k
−1
ES →
k
2
E + P
Michaelis − Menten eq. :
d[P]
dt
= V
m
[S]
[S] + K
m
(1)
If both the substrate (S) and the product (P) are
present, neither can saturate the enzyme. For any
given concentration of S the fraction of S bound to
the enzyme is reduced by increasing the concentra-
tion of P and vice versa. For any concentration of P,
the fraction of P bound to the enzyme is reduced by
increasing concentration of S. When we have S P,
we just have to consider reactions for both directions.
We consider a time discretization of the chemical rate
equation for a reation between a substrate and a prod-
uct with respective stoechiometric coefficient s and p:
s.S → p.P : rate =
1
p
×
d[P]
dt
−→
disc.time
1
p
×
∆[P]
∆T
(2)
(1) and (2) =⇒ p × rate = V
m
[S]
T
[S]
T
+ K
m
≈
[P]
T +timestep
− [P]
T
(T + timestep) − T
We chose to work with a constant timestep:
=⇒ [P]
T +1
= V
m
[S]
T
[S]
T
+ K
m
+[P]
T
(3)
We can note that the Michaelis-Menten constants
(Km) are homogenous to a concentration. We can
then state conc(Km, Level, Time) in our modeling
to set them, where conc stands for concentration.
The experimental response observations of intra-
cellular metabolites to a pulse of glucose were
measured in continuous culture employing automatic
stopped flow and manual fast sampling techniques
in the time-span of seconds and milliseconds af-
ter the stimulus with glucose. The extracellular
Table 1: Concentrations (mM/L) of the Metabolites and
their discretized levels for steady states.
# Metab. Conc. Lvl # Metab. Conc. Lvl
1 glucose 0.055 0 2 g6p 3.480 2
3 f6p 0.600 0 4 fdp 0.272 0
5 gap 0.218 0 6 pep 2.670 2
7 pyr 2.670 2 8 6pg 0.808 1
9 g1p 0.653 0 10 amp 0.955 1
11 adp 0.595 0 12 atp 4.270 2
13 nadp 0.195 0 14 nadph 0.062 0
15 nad 1.470 1 16 nadh 0.100 0
glucose, the intracellular metabolites: glucose-
6-phosphate (g6p), fructose-6-phosphate (f6p),
fructose1-6bisphosphate (fdp), glyceralde-
hyde3phosphate (gap), phospho-enolpyruvate (pep),
pyruvate (pyr), 6phosphate-gluconate (6pg), glucose-
1-phosphate (g1p) as well as the cometabolites: atp,
adp, amp, nad, nadh, nadp, nadph were measured
using enzymatic methods or high performance liquid
chromatography. All the steady-state concentrations
measurements of the E.Coli experiment and their
corresponding discrete levels are summarized in
Table 1.
Inductive Logic Programming, used for induction
or abduction (Mooney, 1997), allows to deal with dis-
crete levels (symbols) and qualitative rules (Doncescu
et al., 2007). Given the background knowledge B and
an observation E (example), the task of ILP is to find
an hypothesis H such that:
• B ∧ H |= E and
• B ∧ H is consistent
Inverse entailment (Inoue, 1992; Muggleton,
1995; Inoue, 2004) enables us to compute H through
deduction by using:
• B ∧ ¬E |= ¬H and
• B 2 ¬H
We are here interested in abducing what happens
during the dynamical transition based on observations
from Table 1.
Inverse entailment for abduction is studied in (In-
oue, 1992) in which abductive computation can be re-
alized by the consequence finding procedure SOL. In
this case, both E and H are sets of literals, so both
¬E and ¬H are clauses. This approach can be further
extended for inducing general hypotheses in (Inoue,
2004), which is generalized from (Muggleton, 1995),
to allow B, E and H for full clausal theories.
SOLAR can be used as an abductive procedure to
infer a hypothesis H in the form of a set of liter-
als. Our logical model is based on the simplified
Michaelis-Menten equation (3) which has here been
represented by three background clauses using the
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
50

conc(Compound, Level, Time) predicate. If we
make the approximations for extreme values in:
[P]
T +1
= V
m
[S]
T
[S]
T
+ K
m
+ [P]
T
(3)
With only 3 levels, as we have in our discretization of
E.Coli experiments, we will get the following simple
rules:
• [S] K
m
⇒
∆[P]
∆T
=
V
m
K
M
⇒ [P]
T +1
= [P]
T
reaction(S,
P, Km) ∧ conc(S, 0, T) ∧ conc(Km, 2, T) ∧
conc(P, L, T)
→ conc(P, L, T+1)
The concentration of the product will not change
between T and T+1 if the reaction is very slow.
• [S] ' K
m
⇒
∆[P]
∆T
=
V
m
2
⇒ [P]
T +1
= V
m
/2 + [P]
T
reaction(S, P, Km) ∧ conc(S, L, T) ∧
conc(Km, L, T) ∧ conc(P, L2, T)
→ conc(P, L2, T+1)
The concentration change of the product between
T and T+1 is not big enough to switch from one
level to another. This is an approximation and a
handy consequence of our discretization (using a
log-scale on real values).
• [S] K
m
⇒
∆[P]
∆T
= V
m
⇒ [P]
T +1
= V
m
+ [P]
T
reaction(S, P, Km) ∧ conc(S, 2, T) ∧
conc(Km, 0, T) ∧ conc(P, L, T)
→ conc(P, 2, T+1)
If the reaction is very quick, it will result in trans-
forming all the substrate into product in one time
step.
If we had more than three levels, we would either need
more rules (they can be automatically generated) or
a general procedure for handling our kinetic model.
This last one is a current implementation issue related
to SOLAR. Another way to deal with more levels be-
ing currently explored consist in the automated gen-
eration of kinetics rules w.r.t. the discretization. Fur-
thermore, we made some simplifications in the path-
ways to be able to use only Michaelis-Menten kinet-
ics, another research topic is to extend our modeling
to reactions ruled by other types of kinetics.
We also added constraints about the unicity of lev-
els at a given time to reduce the number of hypotheses
while keeping consistency:
• ¬conc(S, 0, T) ∨ ¬conc(S, 1, T)
• ¬conc(S, 0, T) ∨ ¬conc(S, 2, T)
• ¬conc(S, 1, T) ∨ ¬conc(S, 2, T)
Now we set the observations for the 6 metabo-
lites (#2 - #7) from Table 1, which have been
possibly affected by the stimulus with glucose,
and the abducibles as those literals of the form
conc( , ,0). Using SOLAR, we get 98 hypothe-
ses as: H76 = conc(g6p,2,0) ∧ conc(adp,2,0)
∧ conc(fdp,0,0) ∧ conc(dhap,0,0) ∧
conc(gap,0,0) ∧ conc(glucose,2,0) ∧
conc(pg3,2,0) ∧ conc(pep,2,0) ∧ conc(atp,0,0)
∧ conc(pyr,2,0)
4 RANKING HYPOTHESES
(Ishihata et al., 2008) (Ishihata et al., 2008) pro-
posed the BDD-EM algorithm that is an implementa-
tion of the expectation maximization algorithm work-
ing on binary decision diagram, allowing it to deal
with boolean functions. (Inoue et al., 2009) (Inoue
et al., 2009) have applied the BDD-EM algorithm to
rank hypotheses obtained through abduction. To rank
our H
1
,... , H
n
hypotheses by probability, we consider
the finite set of ground atoms A that contains all the
values that can take our conc(Compound, Level,
Time) and reaction(Substrate, Product, Km).
Each of the elements of A is a boolean variable.
One of its subsets is the subset of abducibles Γ com-
posed of all the possible values of conc(Compounds,
Level, 0). With θ
i
= P(A
i
) f or A
i
∈ A , we have
to maximize the probability of the disjunction of hy-
potheses helped with the background knowledge B:
F = (H
1
∨ · ··∨ H
n
) ∧ ground(B) to set the good θ pa-
rameters (by the BDD-EM algorithm). F can still be
too big to be retained as a BDD, so an optimisation F
0
of its size is obtained through the use of the minimal
proofs for B and each H
i
. Then, the BDD-EM algo-
rithm computes the probabilities of ground atoms in
A that maximizes the probability of F
0
. Finally, the
probabilities of each hypotheses used for the ranking
are computed as the products of the probabilities of
literals appearing in each H
i
.
To sort our 98 abduced hypotheses, we ran the EM
algorithm on the BDDs corresponding to our hypothe-
ses 10,000 times with random initializations. Note
that if the comparison of these probabilities with each
other is relevant, they should not be taken as abso-
lute probabilities. The 10 most probable abduced hy-
potheses are the following:
These hypotheses are corresponding to our biolog-
ical knowledge that pyruvate is a bottleneck (Peters-
Wendisch et al., 2001) and that the glucose that is to-
tally consumed (e.g. top plot of Fig. 5 from simula-
tion) was in high concentration at the beginning of the
experiment (pulse). It goes along with the very gen-
eral reaction of glycolysis: glucose + 2ADP + 2P +
2NAD
+
→ 2 pyruvate + 2ATP + 2(NADH,H
+
) +
2H
2
O. Also, for some metabolites, such as fructose-
6-phosphate, the levels found through abduction are
KINETIC MODELS AND QUALITATIVE ABSTRACTION FOR RELATIONAL LEARNING IN SYSTEMS
BIOLOGY
51
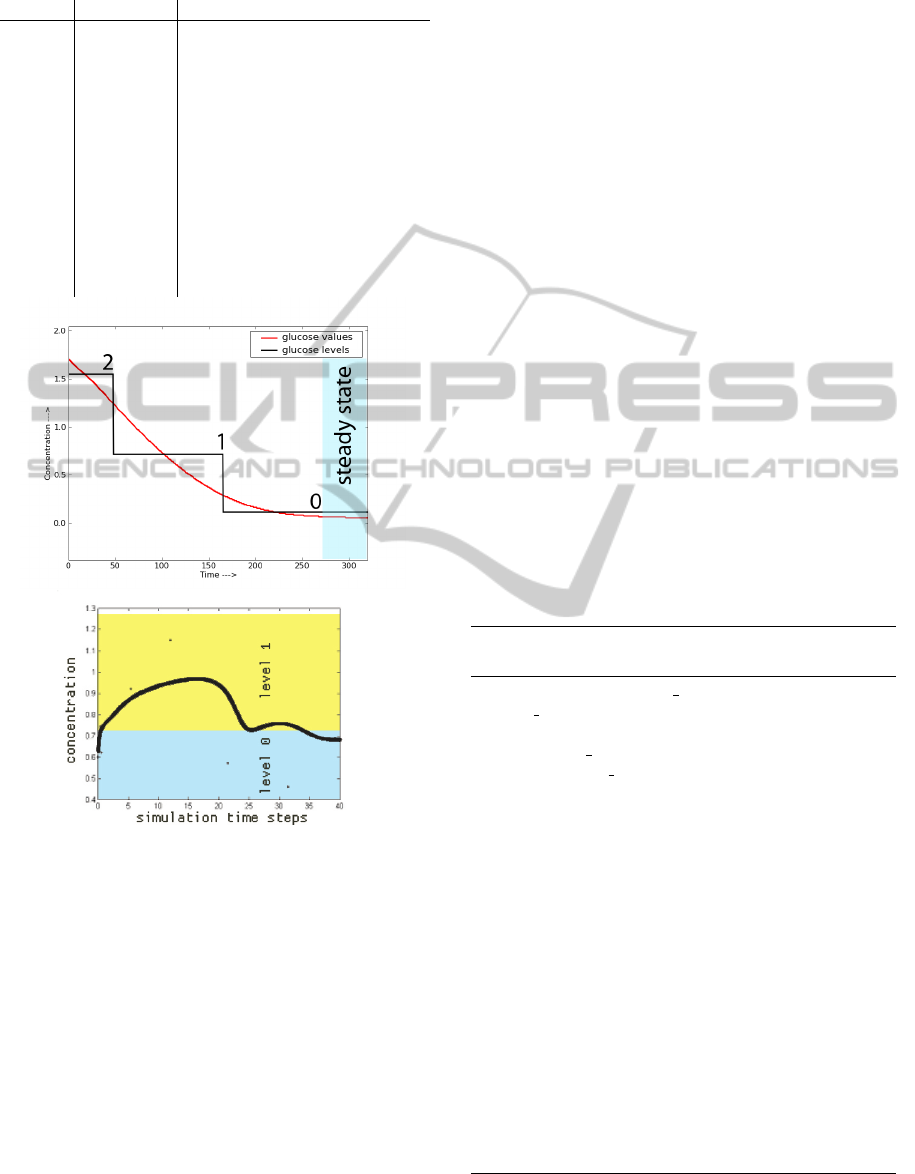
Table 2: 10 Most probable hypotheses.
Hyp. # Probability Abduced conc. levels at T=0
H76 ≈ 1.000 g6p: 2, adp: 2, f6p: 0, fdp: 0,
dhap: 0, gap: 0, glucose: 2,
pg3: 2, pep: 2, atp: 0, pyr: 2
H41 0.822 the same as H76 except pg3: 0
H56 0.625 the same as H76 except g6p: 0
H70 0.553 the same as H76 except atp: 2
H13 0.515 the same as H56 except adp: 0
H90 0.455 the same as H70 except pg3: 0
H82 0.442 the same as H76 except dhap: 2
H43 0.369 the same as H76 except pyr: 1
H9 0.364 the same as H41 except dhap: 2
H68 0.346 g6p: 0, adp: 0, f6p: 0, fdp: 0,
dhap: 0, gap: 0, glucose: 2,
pg3: 2, pep: 2, atp: 2
Figure 5: Top: Discretization of the concentration of glu-
cose in the Glycolysis Pathway of E.Coli after an initial
pulse. Bottom: Simulated evolution of the concentration
of fructose-6-phosphate during the whole experiment.
corresponding to the output of the simulation (e.g bot-
tom plot of Fig.5) with the same low level (0) before
and after the dynamic transition.
5 ENHANCING THE
KNOWLEDGE BASE
Increasing our knowledge about a system is consid-
ered as an iterative process: at first, we consider the
background knowledge combined with the observa-
tions as our knowledge base. Then we produce hy-
potheses and we need to use an algorithm to enhance
(update) our knowledge base with some of the discov-
ered hypotheses, here: abducibles. Ideally, we would
re-run the hypothesis finding process until we can-
not find anything new. This is particularly important
when working with complex chained reactions and
multiple time steps as it can enable deeper learning.
This idea of revising the knowledge base is already
found in (Ray et al., 2009) with a nonmonotonic ap-
proach, but their revision method stays in a qualitative
modeling and do not take quantitative aspects into ac-
count.
Here, it is needed to pick hypotheses that are con-
sistent with the background knowledge and with each
others. For example, if we apply a greedy algorithm
(as Algorithm 1) that picks hypothesis in decreasing
probability order such that the hypothesis add some
knowledge and that our enhanced knowledge stays
consistent, it prevents from abducing other discover-
ables than the ones contained in H76. For instance we
cannot find concentrations at T=0 for ribu5p, rib5p,
sed7p, xyl5p, because if they were abduced, the re-
sulting hypotheses would become inconsistent with
H76. Note also that the abducibles added into the
knowledge base may reduce the computational cost of
later iterations of abduction/induction, but it is com-
parable to discard some branches of exploration.
Algorithm 1: An algorithm to enhance the knowledge
base: most probables firsts.
knowledge ← knowledge base
sorted hypotheses ← sort(hypotheses)
while length(discoverable) > 0 &&
length(sorted hypotheses) > 0 do
tmp ← sorted hypotheses.pop()
if contains(tmp, discoverable) && consistent(tmp,
knowledge) then
knowledge.enhance(tmp)
discoverable.remove(tmp)
end if
end while
With the explicit functions length, pop (destructive), and:
• sort sorts the hypotheses by decreasing probability.
• contains is a function that returns statements of first ar-
gument contained in the second.
• consistent performs consistency checking of two theo-
ries and return True if they are consistent.
• enhance adds statements that are not yet present in the
considered (“self”, “this”) knowledge.
• remove deletes statements from argument present in the
considered (“self”, “this”) object (could make use of
contains).
We could have chosen to pick a combination of hy-
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
52

potheses that discovers more abducibles by penalizing
the solutions including too few different abducibles
with a scoring function inspired by the BIC (Schwarz,
1978): score = −2ln(error) + λ · f (k,n) with k be-
ing the number of chosen hypotheses, n the number
of abducibles, f a function that indicates the struc-
tural complexity of the combination of hypotheses
(decreasing with the increase of n and increasing with
the increase of k) and error the product of the proba-
bilities of chosen hypotheses. We assume here that we
can use their relative significations in error by unbi-
asing the score with a λ parameter. So that the goal of
such an algorithm would be to discover all abducibles
while minimizing this score.
6 CONCLUSIONS
As we found that our results (for time T=0) agreed
with existing background knowledge in biology and
our ODEs-based simulator, this paper showed a
method to deal with the kinetics of metabolic path-
ways with a symbolic model (i.e. Fig 1). We ex-
plained how to discretize biology experiments into
relevant levels to be used with ILP and logic programs
in the large. Moreover, based on these discretization
of concentration into levels, we explained our pro-
cess to transform Michaelis-Menten analytical kinet-
ics equation into logic rules, the authors are not aware
of any previous work in this direction. Therefore the
originality of the work is given by the capacity of a
logical model to find the dynamic response of micro-
organism when a pulse of glucose has been made. We
think that this approach improves the accuracy of the
metabolic flux analysis. Allowing for other kinds of
kinetic modeling (two substrate and/or two products
reactions) would enable us to work with more com-
plete models.
As in (King et al., 2005), this approach tries to
study the behaviour of many ordinary differential
equations while considering a symbolic model with
its advantages whereof the statistical evaluation of hy-
potheses. The process of statistically evaluating hy-
potheses, thanks to BDD-EM (Inoue et al., 2009), is
seen as a good method to find relevant knowledge
among the large quantity of processed data. The prac-
tical validity of this full process (including discretiza-
tion) has been shown by the results of this paper while
working in a well-known theoretical framework (In-
oue, 2004; Mooney, 1997). We strongly believe that
the use of time series discretization and a kinetic mod-
eling to enable ILP to deal with ODE will yield great
results. We also prefer to consider knowledge dis-
covery as an iterative loop where one must review his
knowledge base in the light of new findings (i.e. add
“New KB” next turn in Fig. 2).
Still, our modeling can be improved, and time and
concentration discretization could be finer. Experi-
ments dealing with more than 3 levels and many time
steps will be lead on the Glycolysis and Pentose Phos-
phate pathways of another bacteria, Saccharomyces
Cerevisiae (yeast), with both real world data from
experiments and simulated data. More experiments
with enhancing and updating the knowledge base on
this dataset is necessary to get more accurate results.
A more global approach of discretizing experimental
data and using it in conjunction with automatically
generated symbolic pathways extracted from KEGG
(Kanehisa and Goto, 2000; Kanehisa et al., 2008) can
be applied regardless of the model chosen for infering
new knowledge. This approach can be generically ap-
plied to turn quantitative results from systems biology
into qualitative (symbolic) ones.
REFERENCES
Baral, C., Chancellor, K., Tran, N., Tran, N., Joy, A., and
Berens, M. (2004). A knowledge based approach for
representing and reasoning about signaling networks.
In Proc. of the 12th Int. Conf. on Intelligent Systems
for Molecular Biology, pages 15–22.
Beal, M. (2003). Variational Algorithms for Approximate
Bayesian Inference. PhD thesis, Gatsby Comp. Neu-
rosc. Unit, University College London.
Benhamou, F. (1994). Interval constraint logic program-
ming. Lecture Notes in Computer Science, 910.
Chassagnole, C., Rodrigues, J., Doncescu, A., and Yang,
L. T. (2006). Differential evolutionary algorithms for
in vivo dynamic analysis of glycolysis and pentose
phosphate pathway in Escherichia Coli. A. Zomaya.
Cheeseman, P. and Stutz, J. (1995). Bayesian classifica-
tion (autoclass): Theory and results. In Advances in
Knowledge Discovery and Data Mining, pages 153–
180. The MIT Press.
De Raedt, L. (2008). Logical and Relational Learning.
Springer.
Doncescu, A., Yamamoto, Y., and Inoue, K. (2007). Biolog-
ical systems analysis using Inductive Logic Program-
ming. In IEEE International Symp. on Bioinf. and Life
Science Computing.
Dworschak, S., Grell, S., Nikiforova, V., Schaub, T., and
Selbig, J. (2008). Modeling biological networks by
action languages via answer set programming. Con-
straints, 13(1/2):21–65.
Fages, F., Soliman, S., and France, I. R. (2008). Model
revision from temporal logic properties in systems bi-
ology. In In: Probabilistic Inductive Logic Program-
ming. LNAI, volume 4911, pages 287–304.
KINETIC MODELS AND QUALITATIVE ABSTRACTION FOR RELATIONAL LEARNING IN SYSTEMS
BIOLOGY
53

Gauvain, J.-L. and Lee, C.-H. (1994). Maximum a poste-
riori estimation for multivariate gaussian mixture ob-
servations of markov chains. IEEE Transactions on
Speech and Audio Processing, 2(2):291–298.
Geurts, P. (2001). Pattern extraction for time-series clas-
sification. Lecture Notes in Artificial Intelligence,
2168:115–127.
Inoue, K. (1992). Linear resolution for consequence find-
ing. Artificial Intelligence, 56:301–353.
Inoue, K. (2004). Induction as consequence finding. Ma-
chine Learning, 55:109–135.
Inoue, K., Sato, T., Ishihata, M., Kameya, Y., and
Nabeshima, H. (2009). Evaluating abductive hypothe-
ses using and EM algorithm on BDDs. In Proc. of
IJCAI-09, pages 820–815. AAAI Press.
Ishihata, M., Kameya, Y., Sato, T., and Minato, S. (2008).
Propositionalizing the EM algorith by BDDs. Tech-
nical report, TR08-0004, Dept. Comp. Sc., Tokyo In-
stute of Technology.
Ji, S., Krishnapuram, B., and Carin, L. (2006). Varia-
tional bayes for continuous hidden markov models
and its application to active learning. IEEE Transac-
tions on Pattern Analysis and Machine Intelligence,
28(4):522–532.
Juvan, P., Demsar, J., Shaulsky, G., and Zupan, B. (2005).
Genepath: from mutations to genetic networks and
back. Nucleic Acids Res., 33.
Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa,
M., Itoh, M., Katayama, T., Kawashima, S., Okuda,
S., Tokimatsu, T., and Yamanishi, Y. (2008). KEGG
for linking genomes to life and the environment. Nu-
cleic Acids Res., 36:480–484.
Kanehisa, M. and Goto, S. (2000). Kyoto encyclopedia of
genes and genomes. Nucleic Acids Res., 28(1):27–30.
Keogh, E., Lin, J., and Fu, A. (2005). HOT SAX: efficiently
finding the most unusual time series subsequence. In
5th IEEE International Conference on Data Mining.
King, R., Garrett, S., and Coghill, G. (2005). On the
use of qualitative reasoning to simulate and iden-
tify metabolic pathways. Bioinformatics, 21(9):2017–
2026.
King, R., Whelan, K., Jones, F., Reiser, P., Bryant, C., Mug-
gleton, S., Kell, D., and Olivier, S. (2004). Functional
genomic hypothesis generation and experimentation
by a robot scientist. Nature, 427:247–252.
Kitano, H. (2002). Systems biology toward system-
level understanding of biological systems. Science,
295(5560):1662–1664.
Mooney, R. (1997). Integrating abduction and induction in
machine learning. In Working Notes of the IJCAI97
Workshop on Abduction and Induction in AI, pages
37–42.
Muggleton, S. (1995). Inverse entailment and progol. New
Generation Computing, 13(3/4):245–286.
Nabeshima, H., Iwanuma, K., and Inoue, K. (2003). SO-
LAR: A consequence finding system for advanced
reasoning. In Proc. of the 11th International Con-
ference TABLEAUX 2003, LNAI, volume 2786, pages
257–263.
Peters-Wendisch, P., Schiel, B., Wendisch, V., and et al.,
E. K. (2001). Pyruvate carboxylase is a major
bottleneck for glutamate and lysine production by
corynebacterium glutamicum. Molecular Microbiol.
Biotechnol., 3(2).
Rabiner, L. (1989). A tutorial on hidden markov models
and selected applications in speech recognition. Proc.
of the IEEE, 77(2):257–286.
Ray, O., Whelan, K., and King, R. (2009). A nonmonotonic
logical approach for modelling and revising metabolic
networks. Complex, Intelligent and Software Intensive
Systems, IEEE.
Schwarz, G. (1978). Estimating the dimension of a model.
Annals of Statistics, 6(2):461–464.
Tamaddoni-Nezhad, A., Chaleil, R., Kakas, A., and Mug-
gleton, S. (2006). Application of abductive ILP to
learning metabolic network inhibition from temporal
data. Machine Learning, 64:209–230.
Tiwari, A., Talcott, C., Knapp, M., Lincoln, P., and Lader-
oute, K. (2007). Analyzing pathways using SAT-
based approaches. In Proc of the 2nd Int. Conf. on
Algebraic Biology, pages 155–169.
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
54
