
RECONFIGURABLE COMPUTING IP CORES
FOR MULTIPLE SEQUENCE ALIGNMENT
M. Lakka, A. Desarti, G. Chrysos, E. Sotiriades, I. Papaefstathiou and A. Dollas
Microprocessor and Hardware Laboratory, Department of Electronic and Computer Engineering
Technical University of Crete, Chania, Greece
Keywords: Multiple Sequence Alignment, Reconfigurable Architecture, Bioinformatics.
Abstract: Multiple Sequence Alignment (MSA) is a principal tool in computational molecular biology. MSA is
considered to be a very challenging problem as many software implementations suffer from quadratic time
performance. Two of the best known MSA algorithms, which offer high accuracy and great speed, are T-
Coffee and MAFFT. Reconfigurable technology provides a dramatic reduction of execution time by taking
advantage of high parallelism. It also allows for different problem sizing solutions within a generic
intellectual property (IP) core. This paper presents the implementation of MAFFT and T-Coffee algorithms
on present-day Field Programmable Gate Arrays (FPGAs). The performance of the FPGA systems is
compared against software implementations, concluding that the parallelism of reconfigurable technology
can offer significant computational power to the bioinformatics community.
1 INTRODUCTION
Multiple Sequence Alignment (MSA) organizes a
series of different DNA sequences by finding the
best alignments between them. MSA is a powerful
tool for phylogenetic analysis, protein structure
prediction, homology detection between sequences
and protein family identification. MSA is considered
to be a computationally difficult problem and most
formulations of it lead to NP-complete
combinatorial problems.
As sequence alignment is considered of great
importance in molecular biology, many algorithms
with different features have been proposed and
implemented. Some of the best known are ClustalW
(Thompson et al., 1994), T-Coffee (Notredame et
al., 2000), MAFFT (Katoh et al., 2005), Muscle
(Edgar, 2004), ProbCons (Do et al., 2005) and
Dialign (Morgenster et al., 1998).
Many algorithms give a solution to the MSA
problem but two of them are used most extensively
by the broader community of biologists: MAFFT
(Katoh et al. 2005) and T-Coffee (Notredame et al.,
2000). MAFFT is a method for rapid multiple
sequence alignment based on Fast Fourier
Transform and it offers drastic reduction of the
execution time when compared to other MSA
methods. T-Coffee provides great results in accuracy
at a modest sacrifice in speed.
Reconfigurable computing can offer flexible
architectures with significant performance in various
applications. Specifically, for several bioinformatics
problems many special-purpose architectures have
been proposed. Since the early 90’s more than 10
different research groups have proposed and studied
architectures for all the important sequence
comparison algorithms. Special purpose
reconfigurable architectures have also been proposed
for Phylogenetic trees, RNA and protein structure
prediction, Sequence Homology and Gene Finding.
The community of reconfigurable hardware has
shown great interest in accelerating the process of
multiple sequence alignment. Oliver et al. (Oliver et
al., 2005) used reconfigurable hardware to accelerate
multiple sequence alignment of the ClustalW
algorithm. Xu Lin et al. in (Xu Lin et al., 2005)
analyzed the complexity of ClustalW algorithm and
proposed a strategy to implement efficiently the
algorithm on a Field Programmable Gate Array
(FPGA). Boukerche et al. (Boukerche et al., 2007)
describe the hardware architecture of the most
computationally demanding parts of DIALIGN
algorithm on FPGAs. Shingo Masuno et al. in
(Masuno et al., 2007), developed on an FPGA a
system which implements the Carillo-Lipman
216
Lakka M., Desarti A., Chrysos G., Sotiriades E., Papaefstathiou I. and Dollas A..
RECONFIGURABLE COMPUTING IP CORES FOR MULTIPLE SEQUENCE ALIGNMENT.
DOI: 10.5220/0003167402160221
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2011), pages 216-221
ISBN: 978-989-8425-36-2
Copyright
c
2011 SCITEPRESS (Science and Technology Publications, Lda.)

method. It is obvious that FPGAs offer good
platform for the implementation of systems for
multiple sequence alignment.
The FPGA technology is based on repetitive
patterns of hardware resources (memories, logic,
DSPs) and their interconnection, all of which can be
re-programmed. The proposed architectures of
systems, like those described above, can be either
co-processors that implement the heaviest
computing part of the algorithm, or they can be
stand-alone systems that produce results, or, finally,
they can be intellectual property (IP) cores which are
flexible designs that can be used as a design library.
The main contribution of this work is the
architecture of two IP cores to implement two of the
European Bioinformatics Institute (EBI) preferred
and supported algorithms. This paper presents two
new, parallel architectures of IP cores which
implement the computationally demanding parts of
MAFFT and T-Coffee algorithms. There is an
analysis of the algorithms and a presentation of the
parallel parts that are implemented on an FPGA,
together with the specific advantages of FPGAs vs.
other technologies. As far as the authors know, this
is the first hardware-based approach to execute the
MAFFT and T-Coffee algorithms directly on
hardware.
The rest of this paper is organized as follows:
Section 2 describes the T-Coffee and MAFFT
algorithms and analyzes their nature. Section 3
describes the architecture of the implemented
systems and Section 4 describes the performance for
both implementations. Finally, Section 5 has some
conclusions from this work.
2 MSA ALGORITHMS
AND SOFTWARE ANALYSIS
This section describes the basic steps of the
implemented algorithms, T-Coffee and MAFFT.
2.1 The T-Coffee Method
T-Coffee algorithm is a progressive method for
multiple sequence alignment. T-Coffee works
ideally with datasets of 50 sequences (maximum)
and length 2000 of the sequences (maximum). If the
input is greater than this, the method uses a heuristic
function and it loses its accuracy.
The algorithm comprises of three steps. The first
step constructs a library with the pairwise
alignments between all the input sequences. The
second step extends the library by assigning a
weight for each pair of residue. The final step of the
method implements the progressive alignment
strategy.
2.2 The MAFFT Method
MAFFT algorithm is a progressive method based on
the Fast Fourier Transform (FFT). The MAFFT
method has a new, improved scoring system and a
huge speedup as compared to other existing
methods.
MAFFT consists of three basic steps. Initially, a
distance matrix between all the pairs of input
sequences is constructed. Second, a progressive
guide tree is created from the distances with the
Unweighted Pair Group Method with Arithmetic
Mean. Finally, input sequences are progressively
aligned following the guide tree and using the FFT
algorithm and the normalized scoring system.
2.3 Software Analysis
In order to evaluate the performance bottlenecks, the
software implementations for T-Coffee and MAFFT
were analyzed. Both original software editions of the
algorithms were profiled with the Performance
Analyzer for Linux (Edition 9.1).
The profiling of T-Coffee algorithm showed that
the most computationally demanding part is the
calculation of the alignment score between either
two residues or two lists of residues. The execution
time of this part reaches up to 67% of the total
execution time of the algorithm.
The software analysis of the MAFFT algorithm
showed that the sequence alignment process, which
is the final step of the algorithm, takes up to 80% of
total algorithm execution time.
The software analysis showed that the
implementation of the algorithms’ hotspots on
reconfigurable technology would reduce the total
execution times.
3 IP CORES RECONFIGURABLE
ARCHITECTURES
This section describes the architecture and the
design of the two implemented MSA algorithms.
The implemented architectures can be used in lieu of
the software functions which calculate the alignment
score for the input sequence, thus accelerating the
total performance. The hardware/software
partitioning was modelled with the OSMOSIS tools,
which were developed under the corresponding EU
RECONFIGURABLE COMPUTING IP CORES FOR MULTIPLE SEQUENCE ALIGNMENT
217
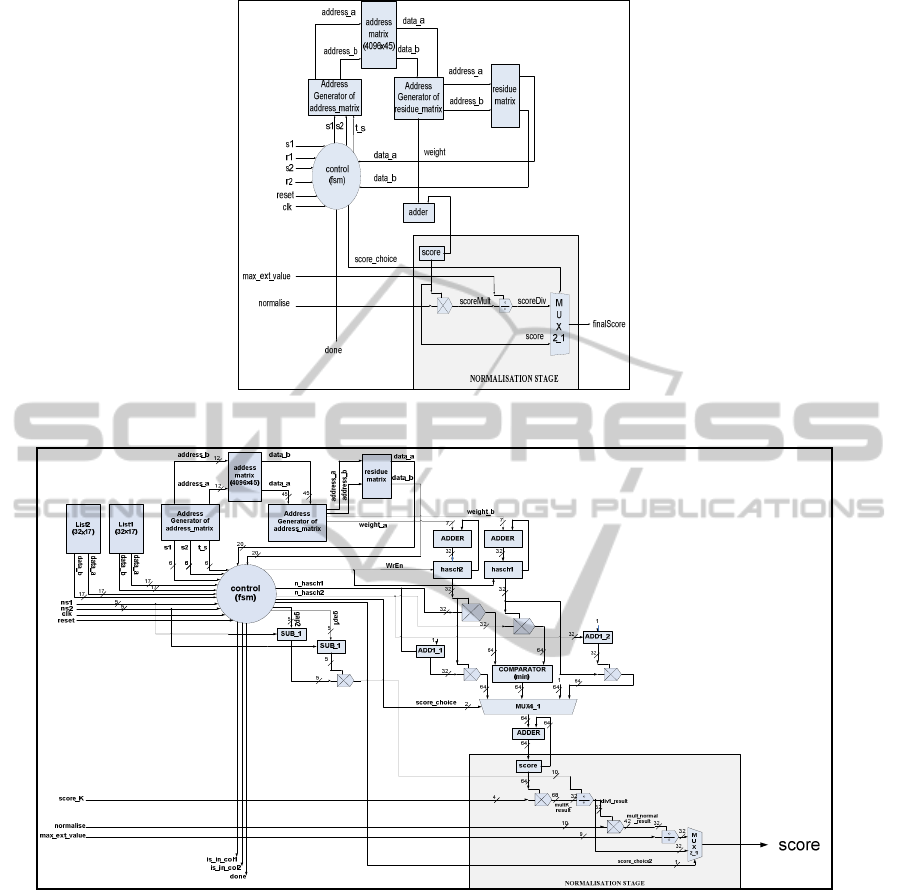
Figure 1: Residue Cost Evaluation Module Architecture.
Figure 2: Constraint List Cost Evaluation Module Architecture.
project (see acknowledgments).
3.1 T-Coffee IP Core Architecture
The architecture of the T-Coffee IP core consists of
two main sub-modules which are used for the two
different parts of the software hotspots. The first
module calculates the alignment score between two
residues, whereas the second one calculates the
alignment score between two lists of residues.
The Residue Cost Evaluation sub-module, shown
in Figure 1, takes as input the IDs of two residues
from different sequences and calculates the score of
their alignment. The calculation of the score is based
on a 3-dimensional lookup table which is pre-loaded
offline to the IP-core for each new input dataset of
sequences. In our architecture the 3D lookup table is
organized into a two-level memory hierarchy.
Fixed-point arithmetic was used for the
normalization stage of the final score, without losing
accuracy vs. the software, as the nature of the
algorithm gives the opportunity of accuracy
reduction without altering the final results.
The Constraint List Cost Evaluation sub-module,
shown in Figure 2, calculates the alignment score
between the residues of two input lists. It takes as
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
218
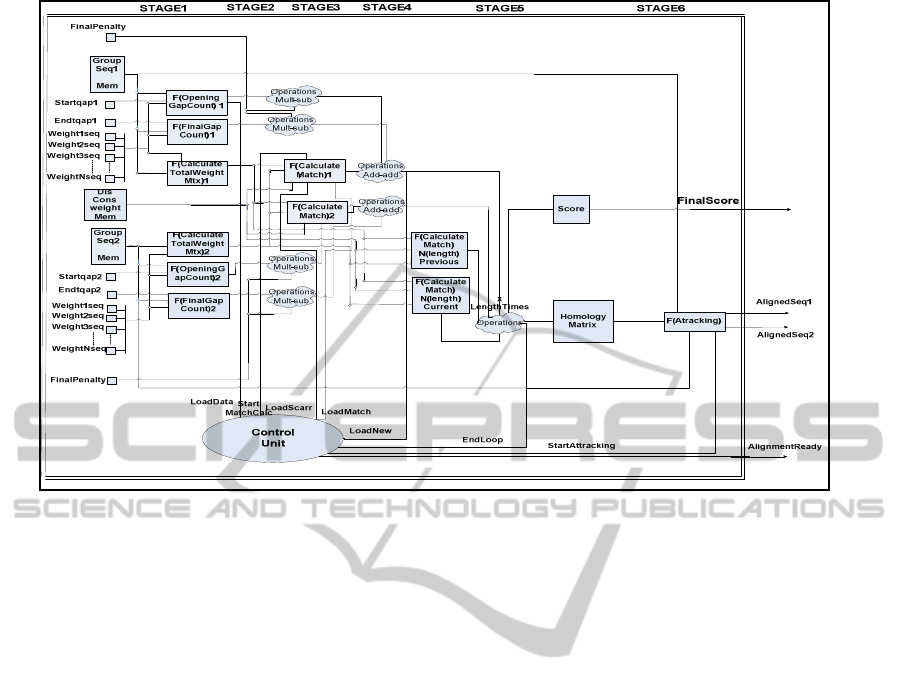
Figure 3: Architecture of MAFFT IP Core.
input two lists of residues from different input
sequences and the numbers of the list elements.
The final score is the total sum of the alignment
scores between all different pairs of the input
residues according to the scoring scheme that was
followed by the Residue Cost evaluation Module.
The normalization stage normalizes the final
score according to the software implementation. The
normalization process, as shown in Figure 2, was
implemented using fixed point arithmetic, with
greater accuracy than the previous module, as the
score in this case is the sum of many different
alignments.
3.2 MAFFT IP Core Architecture
This section describes the architecture of the MAFFT
IP core, shown in Figure 3. The proposed
architecture implements the final step of the MAFFT
algorithm as it takes segments of the input sequences
and outputs the aligned sequences with their
alignment score.
The architecture consists of six-pipelined stages.
The IP core takes as input the weights of the
homologous input sequences and their weights, as
shown in Figure 3. The first four stages calculate the
gap penalties and the weight matrices among the
input sequences. The most complex stage of the
architecture is the fifth one, which calculates the
final alignment score between the aligned sequences
and outputs the final homology matrix of the input
sequences where the sequences are aligned. The
final stage implements the final modification of the
sequence alignment.
Single precision floating point arithmetic was
used for the implementation of all the above
arithmetic structures without losing any accuracy vs.
the software implementation.
Concluding, data transfer is one of major
concerns for these IPs. There exist implementations
of different I/O interfaces giving high data transfer
rates, such as Gigabit Ethernet and PCIe, both of
which are supported by FPGA devices. The transfer
rate for these two interfaces can reach an aggregate
speed of up to 750 MB/sec, which is satisfactory
enough for the implemented algorithms’ demands,
but special care have to be taken in the system that
integrates these IPs in order to fully exploit these
speeds. Thus, the time needed for the data I/O of the
implemented modules is negligible.
4 EXPERIMENTAL RESULTS
This section presents the results of the two IP cores
and compares their performance vs. the original
software implementations of the algorithms. The
performance of software implementations was
computed on an Intel Pentium 4, 2, 66 GHz, with 1
GB RAM and with the Ubuntu 8.04.2 operating
RECONFIGURABLE COMPUTING IP CORES FOR MULTIPLE SEQUENCE ALIGNMENT
219

Table 1: T-COFFEE IP Cores Implementation for Residue Alignment.
Input Sequences
(No of sequences, Sequence
length)
SW Residue Alignment
Execution Time(sec)
FPGA Residue Alignment
Execution Time(sec)
Speedup FPGA vs. Software for
Residue Alignment
(1 IP Core)
Speedup FPGA vs. SW for
indicative parallelism on
Virtex 6 (XCV6SX475T)
1idy_ref2 (22, 65) 2.59 5.76 0.45 x 9.9 x
1wit_ref2 (22, 117) 3.15 10.16 0.31 x 6.8 x
1ag8_ref4 (19, 549) 12.92 323 0.04 x 0.9 x
Table 2: T-COFFEE IP Cores Implementation for Constraint List Residue Alignment.
Input Sequences
(No of sequences, Sequence
length)
SW List Alignment
Execution Time (sec)
FPGA List Alignment
Execution Time (sec)
Speedup FPGA vs. Software for
List Alignment
(1 IP Core)
Speedup FPGA vs. SW for
indicative parallelism
on Virtex 6
(XCV6SX475T)
1idy_ref2 (22, 65) 0.70 3.5 0.2 x 4.4 x
1wit_ref2 (22, 117) 1.08 10.9 0.1 x 2.2 x
1ag8_ref4 (19, 549) 4.29 43.0 0.1 x 2.2 x
Table 3: MAFFT IP Core Implementation.
Input Sequences
(No of sequences,
Sequence length)
SW MAFFT Alignment
Execution Time (sec)
HW MAFFT Alignment
(sec)
Speedup FPGA
vs. Software
(1 IP Core)
Indicative parallelism
on Virtex 6
(XCV6SX475T)
Speedup FPGA vs. SW
for indicative
parallelism on Virtex 6
(XCV6SX475T)
Samplerna (5, 366) 0.007 0.0084 0.83 x 14 11.6 x
flydna10x766(10, 766) 0.10 0.03 3.66 x 14 51.2 x
flydna100x766 (100, 766) 0.54 0.14 3.86 x 14 54.1 x
flydna100x1403 (100, 1403) 1.25 0.24 5.3 x 2 10.6 x
place and route simulated (cycle-accurate
simulation) using the Xilinx ISE 10.1 tool. Their
output results were verified against the original
software.
Both architectures implement the alignment
between two groups of sequences. For the complete
process of the multiple sequence alignment all
possible groups of sequences need to be aligned.
This led us to implement several parallel systems of
IP cores which work independently on alignment
sequences for each input.
4.1 T-Coffee IP Core Results
The T-Coffee IP core was fully designed for a Xilinx
Virtex 4(XC4VFX140) FPGA and it was tested with
three different inputs, as shown in Tables 1 and 2.
The clock rate of the proposed architecture is 146
MHz. The FPGA utilization is very low, offering the
chance of high parallelism. The critical resource of
the implemented IP cores is Block RAMs, and for
the larger datasets the coverage is only 17% of the
total amount of the Block RAMs.
Tables 1 and 2 show i) the software run time for
the calculation of the two alignment scores (residue
alignment and list alignment), ii) the corresponding
time using one IP core, iii) the corresponding
speedups for three different datasets and iv) the
indicative speedup that can be achieved by using a
modern FPGA.
Also, Tables 1 and 2 show that the performance
for single IP core implementation is not impressive;
however, it uses minimal resources. The small
utilization of the FPGA device and the nature of
algorithm can lead to high levels of parallelism. The
alignment of each pair of input sequences can be
computed independently. Taking into account that
large modern devices have a significant number of
Block RAM memories and exploiting data
independence, up to 22 parallel IP cores can operate
in parallel in a single device for both alignment
processes, thus achieving a considerable system-
level speedup, which is more considerable in some
classes of datasets, as described below.
The performance of the implemented system
depends on the size and the nature of the input
sequences. The Block RAM Memories used to store
the input sequences are the bottleneck of the new
architecture, which results in the greater input and
fewer parallel IP cores, and as a result it leads into a
reduction of the performance. As shown in Table 1,
for the very long sequences the system needs high
data retrieval from the memory. This makes the
problem to become control intensive, loosing the
benefits of the hardware parallelism. Finally, the
system was tested with very long sequences which
yielded worse results vs. the software
implementation; however this is not a typical input
dataset as most of the sequences tested in Biology
laboratories have 200-300 residues (max.) length.
4.2 MAFFT IP Core Results
The MAFFT IP core was designed on a Xilinx
Virtex 5 (XC5VSX240T) FPGA. The proposed
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
220

architecture is also very demanding on the number
of Block RAM memories, as for “large” datasets, the
IP core uses the total amount of BRAMs whereas for
“small” datasets, it utilizes about 13% of the
memory. The designed system can process up to
10,000 sequences with 200 residues, each. As shown
in Table 3, the design was tested with six different
kinds of input sequences. The clock rate of this
architecture is 135 MHz for a single IP core. Table 3
shows the run times for all the measured input
sequences for a general purpose processor running
original software and for the designed IP core and
the perspective speedup. For the “large” datasets IP
Core is 4 to 5 times faster. For the “small” datasets
things are not that good but following the
considerations that we made for the T-Coffee IP
core, for the “small” datasets of MAFFT we can
assume that for a large modern FPGA device we can
have up to 15 parallel MAFFT IP cores, thus
achieving speedup from 10 to 55 times vs. a high
end general purpose processor.
These IP cores can be set up for different sizes of
datasets, which makes reconfigurable computing
preferable to VLSI due to the resulting flexibility to
“tune” the design to the dataset type.
5 CONCLUSIONS
Two of the five best known algorithms for multiple
sequence alignment implemented and used by the
European Bioinformatics Institute (EBI) are the
MAFFT and T-Coffee algorithms. This work
presents FPGA technology-based IP cores for the T-
Coffee and MAFFT algorithms. This is to the
authors’ knowledge the first work in the literature in
which there is an attempt to model these two
algorithms in reconfigurable hardware. Experimental
results show that reconfigurable technology can
offer significant performance boosting, especially in
cases in which the input data allows for high
parallelism. Future research will focus on
performance improvement of the designed IP cores
by increasing the number of parallel machines. As
internal memory (BRAMS) is the critical resource,
storing the input sequences in external memory
(DDR) can free the internal memory for more
parallel machines. The hardware integration of the
designed IP cores with the rest of the algorithm
running in software can lead to systems that can be
used by biologists.
ACKNOWLEDGEMENTS
This publication is based on work performed in the
framework of the FP7 Project OSMOSIS, which is
funded from the European Community’s Seventh
Framework Programme(FP7/2007-2013) under grant
agreement FP7-SME-222077.The authors would like
to acknowledge: the contributions to the OSMOSIS
Project of their colleagues in Algosystems SA,
Electronic Design Ltd, Dunvegan, Politecnico di
Torino, CEA, and TSI.
REFERENCES
Thompson, J. D., Higgins, D. G., Gibson, T. J., 1994.
CLUSTAL W: improving the sensitivity of
progressive multiple sequence alignment through
sequence weighting, position-specific gap penalties
and weight matrix choice. Nucleic Acids Research, vol
22, pp. 4673-4690.
Notredame, C., Higgins, D. G., Heringa, J., 2000. T–
Coffee: A novel method for fast and accurate multiple
sequence alignment. Journal of Molecular Biology,
vol. 302, issue 1, pp. 205-217.
Katoh, K., Kuma, K., Toh, H., Miyata, T., 2005. MAFFT
version 5: improvement in accuracy of multiple
sequence alignment. Nucleic Acids Research, vol 33,
pp. 511-518.
Edgar, R. C., 2004. MUSCLE: a multiple sequence
alignment method with reduced time and space
complexity. BMC Bioinformatics, 5:113.
Do, C. B., Mahabhashyam, M. S. P., Brudno, M.,
Batzoglou, S., 2005. PROBCONS: Probabilistic
Consistency-based Multiple Sequence Alignment.
Genome Research, vol. 15, pp. 330-340.
Morgenster, B., French, K., Dress, A., Werner, T, 1998.
DIALIGN: Finding local similiraties by multiple
sequrnce alignment. Bioinformatics, vol. 14, No. 3, pp.
290-294.
Oliver, T., Schmidt, B., Nathan, D., Clemens, R., Maskel,
D., 2005. Using reconfigurable hardware to accelerate
multiple sequence alignment with ClustalW.
Bioinformatics, vol. 21, No. 16500, pp. 3431-3132.
Lin, X., Peiheng, Z., Dongbo, B., Shengzhong, F.,
Ninghui, S., 2005. To accelerate Multiple Sequence
Alignment using FPGAs. High-Performance
Computing in Asia-Pacific Region, pp. 176-180.
Boukerche, A., Correa, J. M., Melo, A. C. M. A., Jacobi,
R. P., Rocha, A. F., 2007. An FPGA-based accelerator
for multiple biological sequence alignment with
DIALIGN. International Conference on High
Performance Computing, pp. 71-82.
Masuno, S., Maruyama, T., et al., 2007. An FPGA
Implementation of Multiple Sequence Alignment
Based on Carrillo-Lipman Method. In Proceedings of
Field Programmable Logic and Applications, pp. 489-
492.
RECONFIGURABLE COMPUTING IP CORES FOR MULTIPLE SEQUENCE ALIGNMENT
221
