
A CLOUD ARCHITECTURE FOR BIOINFORMATICS
WORKFLOWS
Hugo Saldanha, Edward Ribeiro, Maristela Holanda, Aleteia Araujo
Genaina Rodrigues, Maria Emilia Walter, Jo˜ao Carlos Setubal and Alberto D´avila
Department of Computer Science, University of Brasilia, Brasilia, Brazil
Virginia Bioinformatics Institute, Blacksburg, U.S.A.
Institute Oswaldo Cruz, FIOCRUZ, Rio de Janeiro, Brazil
Keywords:
Cloud architecture, Bioinformatics workflow, High-throughput genome sequencing.
Abstract:
Cloud computing has emerged as a promising platform for large scale data intensive scientific research, i.e.,
processing tasks that use hundreds of hours of CPU time and petabytes of data storage. Despite being object
of current research, efforts are mainly based on MapReduce in order to have processing performed in clouds.
This article describes the BioNimbus project, which aims to define an architecture and to create a framework
for easy and flexible integration and support for distributed execution of bioinformatics tools in a cloud envi-
ronment, not only tied to the MapReduce paradigm. As a result, we leverage cloud elasticity, fault tolerance
and, at the same time, significantly improve the storage capacity and execution time of bioinformatics tasks,
mainly of large scale genome sequencing projects.
1 INTRODUCTION
Cloud computing has emerged as a promising plat-
form for large scale data intensive computation. Its
ability to provide a flexible and on-demand comput-
ing infrastructure with large scalability enables the
distribution of the processing among a large number
of computing nodes, that considerably reduces the ex-
ecution time due to task parallelization.
Certain types of scientific workflows need to
produce and analyze an enormous amount of data,
frequently with the addition of complex calcula-
tions. Bioinformatics area is a proeminent exam-
ple of such domain. Improvements in the tech-
niques of genome sequencing due to the new gen-
eration high-throughput sequencing machines (Illu-
mina, 2010) produce a large volume of data, order
gigabytes, that must be analyzed in a sequencing
project (Benson et al., 2009).
Cloud computing is interesting to both academia
and industry communities, mainly because it reduces
costs associated to the management of hardware and
software resources in an out-sourced and pay-per-use
basis. A cloud environment can successfully provide
resources for these bioinformatics requirements. In
fact, some projects, such as Cloudburst(Schatz, 2009)
and Crossbow (Langmead et al., 2009), have taken ad-
vantage of cloud computing environments. However,
these projects have only addressed parts of the bioin-
formatics workflow. A more generic and flexible ap-
proach is needed if non trivial workflows are to be
applied to cloud environments.
In this context, this paper proposes a service ori-
ented cloud architecture to be used for bioinformatics
tasks, specially in workflows in the context of high-
throughput genome sequencing projects. We believe
that this environment can deal with the enormous
amount of data produced by those projects. Besides,
this work in progress has the objectives of improving
usability and to use commodity hardware. Therefore,
a large number of biologists and organizations, espe-
cially in developing countries, will be able to share
their computational resources and to easily use cloud-
based technology in their projects.
This paper is organized as follows. First, in Sec-
tion 2 we briefly describe the characteristics of high-
throughput genome sequencing projects. Section 3
shows how cloud computing has been applied in such
projects. In Section 4 we propose a cloud-based ar-
chitecture for bioinformatics workflows, stressing our
contributions. Finally, in Section 5, we conclude and
point the next steps.
477
Saldanha H., Ribeiro E., Holanda M., Araujo A., Rodrigues G., Emilia Walter M., Carlos Setubal J. and Dávila A..
A CLOUD ARCHITECTURE FOR BIOINFORMATICS WORKFLOWS.
DOI: 10.5220/0003394004770483
In Proceedings of the 1st International Conference on Cloud Computing and Services Science (CLOSER-2011), pages 477-483
ISBN: 978-989-8425-52-2
Copyright
c
2011 SCITEPRESS (Science and Technology Publications, Lda.)
2 HIGH-THROUGHPUT
GENOME SEQUECING
PROJECTS
Sequencing is the task of discovering the bases (ade-
nine, cytosine, guanine and thymine) forming the
DNA (chromosomes) of one or more organisms.
Then, in a very general view, a genome sequencing
project is developed by a team composed of biolo-
gists, in the molecular biology laboratories, and com-
puter scientists, in computer laboratories, with the ob-
jective of reconstructing the DNA of the studied or-
ganism(s), since the automatic sequencing machines
can not sequence large fragments of DNA.
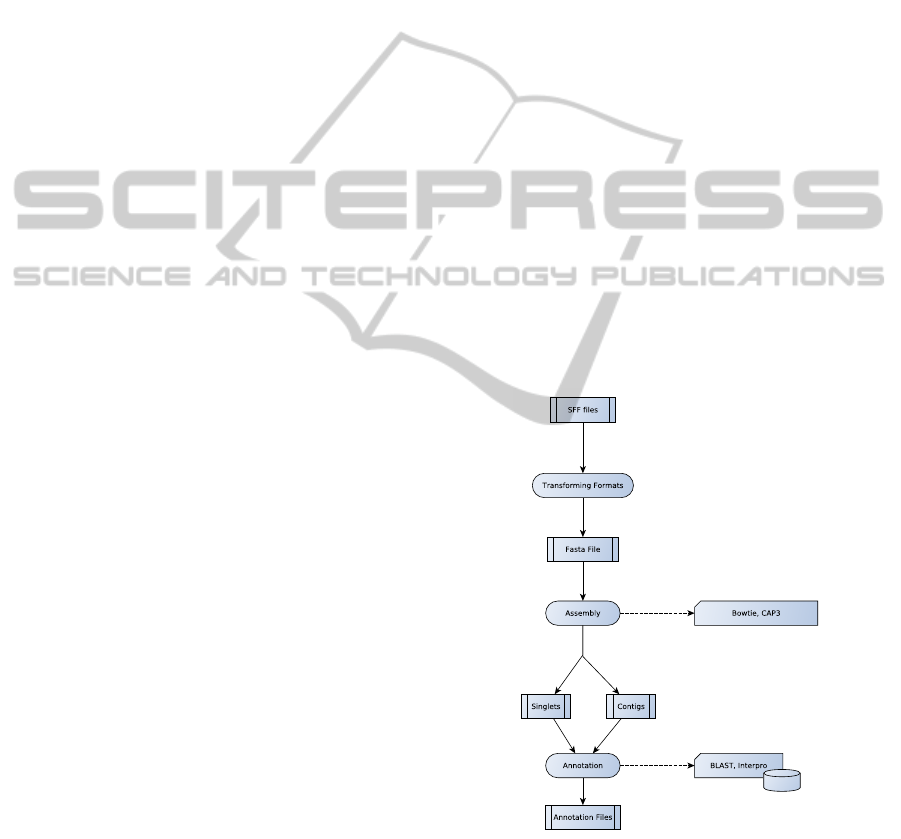
A bioinformatics workflow (pipeline) for large
scale genome sequence project can be divided into
two or three phases, depending on the objectives of
a particular project: assembly, mapping, and annota-
tion (Figure 1), where the output of each phase is the
input of the next one. A workflow can be constructed
with the phases: assembly and annotation, mapping
and annotation or assembly, mapping and annotation.
A large number of short sequences of DNA are
produced by the automatic sequencers and trans-
formed into strings having letters in the alphabet Σ =
{A, C, G, T}, corresponding to the four DNA bases.
This is done in the biology laboratories. The fol-
lowing tasks are done in the bioinformatics labora-
tory. The assembly phase groups fragments of DNA
sharing similar extremities, in order to reconstruct the
original DNA sequence. Files conataining contigs
(a group composed of two or more fragments repre-
sented by a sequence obtained by the consensus of
its fragments), singlets (fragments that could not be
grouped) and other auxiliary files are the output of this
phase. The mapping phase identifies the localizations
of the short sequences inside a reference genome, and
possibly generates groups containing one or more se-
quences. The last step, annotation, aims to discover
biological functions to the groups constructed in the
previous phases, comparing its sequences with other
sequences already stores in databases of proteins and
non-coding RNAs, or try to identify unknown genes
and non-coding RNAs, among other functions.
DNA sequecing has seen great improvements
since the rise of new technologies developed by Illu-
mina (Illumina, 2010), Applied Biosystems (Biosys-
tems, 2010) and 454 Life Sciences (Sciences, 2010).
This next-generation of high-throughput sequenc-
ing machines is capable of sequencing millions of
short sequences of DNA called reads from the target
genome in a single run. Each of these reads contain
from 25 base-pairs up to hundreds of them.
The repetitive execution of a large volume of in-
terdependent tools, besides management tasks of data
flow, turn workflows excessively complex and time
consuming. Management software of workflows has
the following operational requirements:
• High throughput: the system should be able
to handle large datasets, complex data analysis
workflows and large numbers of jobs requiring
long periods of processing time.
• Ease-of-use: well designed GUI that makes the
workflow easy and intuitive to use by non-experts
end-users.
• Flexibility: make it ease to include new and up-
dated tools in the workflow, so that the system
presents modularity and flexibility;
• Modularity: it should be easy for the operator
to track changes in biological databases and its
affected parts of the workflow so that it can re-
execute only the affected parts with minimal re-
dundancy.
There are a number of workflow systems publicly
available as Cyrille2 (Cyr, 2008) and Taverna (Hull
et al., 2006). Each workflow provides complementary
sets of features, but to the best of our knowledge, none
of them is designed for a cloud-based environment.
Figure 1: An example of a bioinformatics workflow with
assembly and annotation phases.
Now we discuss two examples to show the amount
of data that must be analysed in genomeprojects. (Fil-
ichkin et al., 2010) worked with approximately 271
million of 32 base-pairs reads sequenced using the Il-
lumina technology. They mapped the reads of a small
CLOSER 2011 - International Conference on Cloud Computing and Services Science
478

genome (about 120M base-pairs) to the Arabidopsis
thaliana genome, in order to identify alternativesplic-
ing. In order to find alternative splicing in the hu-
man genome, two groups of researchers (Sultan et al.,
2008; Pan et al., 2008) mapped about 15 millions of
reads to the whole human genome, which consists of
approximately 3 Gb of information only in this map-
ping phase.
In summary, after the generation of enormous
amounts of sequence data by the automatic se-
quencers, these short reads must be analysed using
many different bioinformatics techniques that are ex-
tremely computer resources consuming, for example,
comparison of genomes of closely related species, de-
tection of genomic variations among human chromo-
somes, identification of differentialy expressed genes
and finding new alternative splicing of genes.
3 CLOUD COMPUTING FOR
BIOINFORMATICS
In this section we first present main characteristics of
cloud computing and after we describe related works
of clouds and bioinformatics.
3.1 Cloud Computing Technologies
A large cloud computing ecosystem consisting of
technologies and service providers creates a complex
environment that provides a plethora of choices for its
users. Thus, proposing an architecture for the cloud
computing environment that encompasses all possi-
bilities without losing its generality has imposed a
great deal of efforts to researchers.
The first problem is the cloud definition itself. As
one tries to establish its own definition of cloud, of-
ten just one or few aspects are focused, making it
not complete for other cases. Gonzalez et al. (Gon-
zalez et al., 2009) try to address this situation, gather-
ing different definitions, in hope that commonalities
could be found. The conclusion is that there is not
any definition yet, but it is possible to characterize the
mininum requirements for a cloud environment: scal-
ability, pay-per-use utility model and virtualization.
Usually, the cloud computing environment is pre-
sented as a stack, where every layer encompasses a
type of service. There are three layers that are de-
fined when a cloud stack: Infrastructure-as-a-Service
(IaaS), Platform-as-a-Service (PaaS) and Software-
as-a-Service (SaaS) (Gonzalez et al., 2009; Lenk
et al., 2009; Hayes, 2008).
A known programming model for cloud comput-
ing is the Map-Reduce (Dean and Ghemawat, 2010),
developed by Google. Microsoft Dryad is a gener-
alization of the Map-Reduce data flow framework.
This model is ubiquitous and has been increasingly
used in the academic community and industry through
its most famous open source implementation, called
Hadoop (White, 2009). In this model, processing is
divided into two phases: map and reduce. During the
map phase, key-value pairs are computed from the in-
put files. Then, these data are shuffled, sorted and
automatically grouped by the key. Finally, in the re-
duce phase, all values grouped by a key are processed
together, producing an output. In spite of its popular-
ity, Hadoop’s Map-Reduce has a number of limiting
factors as the following:
• Map-Reduce model is inflexible in that all the pro-
cessing needs to be divided into two steps, e.g., a
Map and a Reduce step. The consequences of this
approach are two fold: when the processing is rel-
atively simple, one of the steps (usually reduce) is
unnecessary, but it still needs to be performed by
the framework generating more overhead and in-
creasing the total execution time. Secondly, com-
plex processing may require more than one Map-
Reduce job, and this further increases the com-
plexity of the task.
• Hadoop Map-Reduce cannot leverage hardware
heterogeneity and, therefore, make use of a lim-
ited set of task scheduling algorithms among com-
puting nodes;
• The Map-Reduce framework stores all its inter-
mediate results on disk, increasing the total exe-
cution time;
• The shuffle/sort step is always executed even
when it is not necessary;
• Map-Reduce is essentially a batch style model of
computing, in spite of current work that aims to
adapt it to interactive execution (Ekanayake et al.,
2010).
In spite of these limitations, Map-Reduce has a
simple distributed programming model and leverages
the use of commodity hardware so we hope to provide
a cloud architecture that can be able to seamless inte-
grate with Map-Reduce, but also fill the gap left by
the Map-Reduce model of computation, that is, to al-
low more flexible and efficient models of distributed
execution.
3.2 Bioinformatics Cloud Applications
and Related Work
Applications like Cloudburst (Schatz, 2009) and
Crossbow (Langmead et al., 2009) are examples of
A CLOUD ARCHITECTURE FOR BIOINFORMATICS WORKFLOWS
479

bioinformatics tools that run in the cloud environ-
ment, using Hadoop’s Map-Reduce. Cloudburst is
a parallel Map-Reduce algorithm optimized for map-
ping high-throughputgenome sequence reads to other
reference genomes, using a variety of biological anal-
yses. Crossbow is a Hadoop-based software tool that
combines a sequence aligner and a SNP caller us-
ing the Map-Reduce model to parallelize the pro-
cessing of inputs in large scale. Both applications
are allowed to run either on a private cloud or on a
public cloud like Amazon’s Elastic Compute Cloud
(EC2) (Inc, 2008) computing service. Microsoft’s
Dryad and Azure were also applied for bioinformatics
applications (Qiu et al., 2009), significantly increas-
ing their performance.
In this direction, some efforts have been made
to provide an easy-to-use architecture that facilitates
researchers to compose and execute applications in
bioinformatics workflows. One example is the inte-
gration (Wang et al., 2009) of Hadoop’s Map-Reduce
implementation with Kepler (Altintas et al., 2004), a
scientific workflow management system. The middle-
ware Hydra (Ogasawara et al., 2009) has been used to
achieve data parallelism in bioinformatics cloud ap-
plications coupled to provenance facilities (Coutinho
et al., 2010). Although these applications use the
cloud computing power, they are aimed at solving
specific bioinformatics problems, not trying to be
a complete solution to the applicability of complex
workflows in the cloud.
4 A CLOUD ARCHITECTURE
FOR BIOINFORMATICS
WORKFLOWS
Two of the examples cited in sub-section 3.2 are pri-
marily based on Hadoop to solve their specific tasks,
and this represents a trend in cloud based systems.
Cloud computing is an alternative feasible for other
problems in bioinformatics. In this section, we pro-
pose BioNimbus, a cloud architecture that allows the
parallel execution of algorithms and efficient strate-
gies for storage of genome datasets. Our architecture
(Figure 2) occupies the PaaS layer of the cloud stack
and the architecture itself is composed by controllers
that allow the provisioning and execution of bioinfor-
matics applications in a distributed and flexible way.
Our design is centered on a Service Oriented Ar-
chitecture (SOA) where each bioinformatics appli-
cation as BLAST (Altschul et al., 1990) or Inter-
pro (Mulder et al., 2007), for example, can be made
available as a web service. In fact, bioinformatics
workflows as Biomoby (Wilkinson and Links, 2002)
already use SOAP based web services for remote ex-
ecution of bioinformatics tools. But we opted for
RESTful Web Services (Richardson and Ruby, 2007)
because it eliminates much of the complexity of de-
veloping and maintaining distributed SOAP services
and providing more flexible interfaces based on URL
and HTTP verbs as GET, PUT, DELETE and POST.
Each service will have a logical identifier that is
mapped to one or more instances of services.
From top down, the first layer is the entry point to
user access, and it provides both an interactive web
frontend, as well as a, RESTful endpoint so that this
layer allows the user to interact with the cloud ser-
vices from both an interactive or programmatic inter-
face. The second layer is the core of our architec-
ture, and it is composed of a set of controllers, namely
the Service Manager, Monitoring Manager, Job/Task
Manager, and Resource Manager. It is important to
note that we are designing the pluggable interfaces so
that more managers can be added to the core engine.
The roles of each manager are defined as follows:
• Service Controller: responsible for service regis-
tration, configuration, discovery, monitoring and
invocation;
• Job/Task Controller: responsible for instantiation
of jobs and tasks on worker machines;
• Provisioning Controller: responsible for the allo-
cation of idle machines and load balancing among
multiple instances of service, and increasing or
decreasing the number of instances of a particu-
lar service, as it becomes more or less requested;
• Monitoring Controller: responsible for collecting
statistics about the health and status of machines
in the cloud.
The next layer is the Infrastructure-as-a-Service
(IaaS), that includes the local execution of tools, as
well as, the Hadoop suite and virtualization environ-
ment where the software stack is executed.
The data generated by each instance will be per-
sisted in a distributed storage as HDFS or Cassan-
dra, which automatically replicates and balance data
across machines. Each manager has a embedded
HTTP server that allows the user to invoke actions
by the means of a RESTful API. One of the chal-
lenges of this architecture is to enable service compo-
sition in a quick and intuitive way, allowing the cre-
ation of optimized work in a data flow processing in
large scale. Finally, each Instance Worker actually ex-
ecutes and monitors the individual executions of data
intensive tasks as running BLAST on a large dataset
in each single host. As with Map-Reduce and Dryad,
re-execution is used to cope with failures of individual
CLOSER 2011 - International Conference on Cloud Computing and Services Science
480

nodes. We also hope to enable the auto-provisioning
and composability of such tools in a intuitive manner.
Graphical User Interface
MAP−REDUCE
HADOOP
HBASE
HDFS
Local Service
PaaS
IaaS
Middleware
Job/Task Manager
Service Manager
(Peer−to−Peer)
Resource Manager
Monitoring Manager
Java Virtual Machine
Operating System
Virtual Machine
Figure 2: A cloud architecture for bioinformatics work-
flows.
Our architecture is composed by a set of indepen-
dent components that communicate through the net-
work to provide and consume services. This poses
two main challenges: firstly, we need to avoid single
points of failure, so our architecture is based on Peer-
to-Peer(P2P) technology, particularly the use of an
implementation of the Kademlia (Stoica et al., 2003),
so that services can discover one another and route
messages in a totally decentralized way. Distributed
Hash Tables (DHTs) like Chord (Stoica et al., 2003)
and Kademlia (Maymounkov and Mazieres, 2002)
proved to be appropriate for the deployment of re-
silient cloud services, as evidenced by Amazon’s Dy-
namo system (DeCandia et al., 2007) and Apache
Cassandra (Lakshman and Malik, 2009). Another
reason to use a DHT is to enable the communication
between data centers, a requirement of modern cloud
computing environments. Our architecture should in-
tegrate seamless with Hadoop tools (HDFS, HBase,
Zookeeper and Map-Reduce) as well as with other
cloud-friendly storage systems like Apache Cassan-
dra, for example.
The following scenario details a possible use of
the proposed architecture as depicted in Figure 3:
1. The user logins on the web frontend;
2. The user performs a look up of the services to
be executed (Bowtie and BLAST) in the Service
Controller;
3. The user uploads a set of fasta files to the dis-
tributed file system;
4. The user dispatches the job where the first task
runs Bowtie followed by the execution of BLAST
on the resulting files to the Job/Task Controller;
5. The Job/Task Controller validates both the input
format file and the service availability and addi-
tional parameters passed informed;
6. The Job/Task Controller contact the Provisioning
Controller that allocates a set of idle machines
according to load, CPU, and storage capacities,
scheduling a machine for each input file;
7. Each file is submitted to a single machine where it
is processed by Bowtie and the results are stored
on a distributed file system;
8. As soon as each output file is produced by Bowtie,
the BLAST is executed on the same machine so
that it can take advantage of data locality;
9. The results of Bowtie and BLAST execution are
stored on a distributed file system as well as log
and statistics files;
10. The user is able to inspect and retrieve the data
files produced by the execution of the two tools,
Bowtie and BLAST;
The execution flow just illustrated enables to com-
pose and execute cloud services that may or may not
include map-reduce jobs in an interactiveand intuitive
manner. We believe that future cloud services should
enable this to be used in a bioinformatics context.
5 CONCLUSIONS
In this paper we described our current work towards
a distributed, resilient, cloud-based architecture to be
used in bioinformatics workflows. In spite of its
native integration with Hadoop’s Map-Reduce, we
are starting to explore other cloud based program-
ming models and how they can be influenced by
the unique requirements of large scale sequencing
genome projects.
The ultimate goal of this project is to enable the
management of bioinformatics tools in a cloud based
environment, but its architecture is being conceived
in such a way that different models in IaaS and PaaS
could be integrated to it in a straightforward manner.
We plan to investigate storage backends as Hadoop’s
HBase and Cassandra as better alternatives to store
biological data in a consistent, replicated, and fault
tolerant way in the cloud.
A CLOUD ARCHITECTURE FOR BIOINFORMATICS WORKFLOWS
481
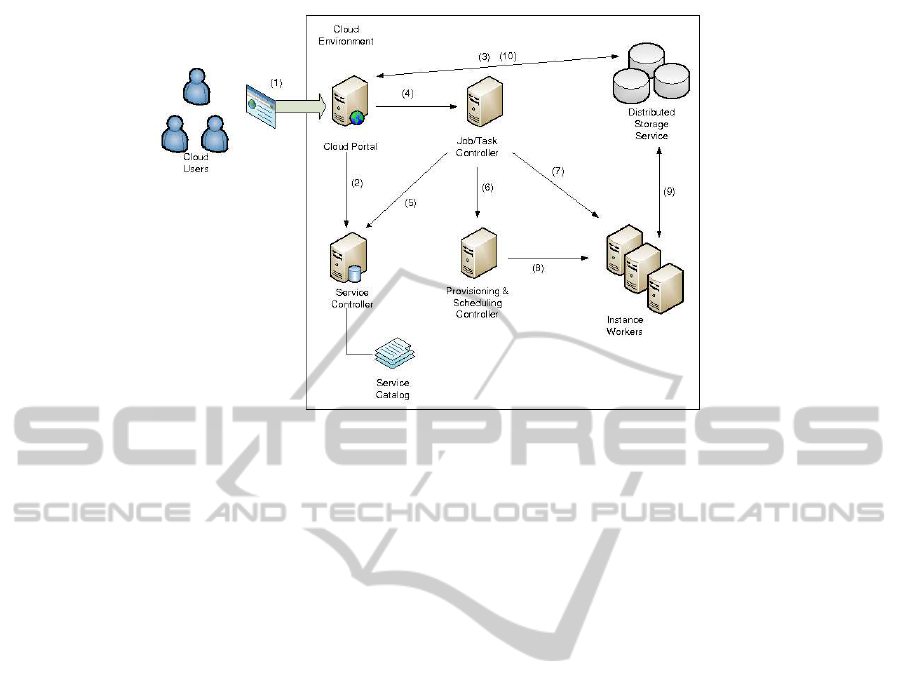
Figure 3: The flow of execution of a bioinformatics application in the proposed cloud architecture.
Next, we plan to implement our BioNimbus cloud
using a Peer-to-Peer plaform based on Kademlia, pro-
pose an efficient data storage for sequences produced
by high-throughput automatic sequencer and for the
results of the execution of Bowtie and BLAST. Be-
sides, we plan to investigate how workflows of bioin-
formatics could be efficiently used in our cloud.
REFERENCES
(2008). High-throughput bioinformatics with the Cyrille2
pipeline system. BMC bioinformatics, 9(1):96+.
Altintas, I., Berkley, C., Jaeger, E., Jones, M., Ludascher,
B., and Mock, S. (2004). Kepler: An extensible sys-
tem for design and execution of scientific workflows.
In Proceedings of the 16th International Conference
on Scientific and Statistical Database Management,
pages 423–, Washington, DC, USA. IEEE Computer
Society.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and
Lipman, D. J. (1990). Basic local alignment search
tool. Journal of molecular biology, 215(3):403–410.
Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell,
J., and Sayers, E. W. (2009). GenBank. Nucleic acids
research, 37(Database issue):D26–31.
Biosystems, A. (2010). Applied Biosystems.
http://www.appliedbiosystems.com.
Coutinho, F., Ogasawara, E., de Oliveira, D., Braganholo,
V., Lima, A. A. B., D´avila, A. M. R., and Mattoso,
M. (2010). Data parallelism in bioinformatics work-
flows using hydra. In Proceedings of the 19th ACM
International Symposium on High Performance Dis-
tributed Computing, HPDC ’10, pages 507–515, New
York, NY, USA. ACM.
Dean, J. and Ghemawat, S. (2010). Mapreduce: A flexible
data processing tool. Communications of the ACM,
53(1):72–77.
DeCandia, G., Hastorun, D., Jampani, M., Kakulapati,
G., Lakshman, A., Pilchin, A., Sivasubramanian, S.,
Vosshall, P., and Vogels, W. (2007). Dynamo: Ama-
zon’s highly available key-value store. SIGOPS Oper.
Syst. Rev., 41(6):205–220.
Ekanayake, J., Li, H., Zhang, B., Gunarathne, T., Bae, S.-
H., Qiu, J., and Fox, G. (2010). Twister: A Runtime
for Iterative MapReduce.
Filichkin, S. A., Priest, H. D., Givan, S. A., Shen, R.,
Bryant, D. W., Fox, S. E., Wong, W., and Mockler,
T. C. (2010). Genome-wide mapping of alternative
splicing in arabidopsis thaliana. Genome Research,
20(1):45–58.
Gonzalez, L. M. V., Rodero-Merino, L., Caceres, J., and
Lindner, M. (2009). A break in the clouds: towards a
cloud definition. SIGCOMM Computer Communica-
tion Review, 39(1):50–55.
Hayes, B. (2008). Cloud computing. Communications of
the ACM, 51:9–11.
Hull, D., Wolstencroft, K., Stevens, R., Goble, C., Pocock,
M., Li, P., and Oinn, T. (2006). Taverna: a tool for
building and running workflows of services. Nucleic
Acids Research, 34(Web Server issue):729–732.
Illumina (2010). Illumina Inc. http://www.illumina.com.
Inc, A. (2008). Amazon Elastic Compute
Cloud (Amazon EC2). Amazon Inc.,
http://aws.amazon.com/ec2/#pricing.
Lakshman, A. and Malik, P. (2009). Cassandra: structured
storage system on a p2p network. In Proceedings of
the 28th ACM symposium on Principles of distributed
computing, PODC ’09, pages 5–5, New York, NY,
USA. ACM.
CLOSER 2011 - International Conference on Cloud Computing and Services Science
482

Langmead, B., Schatz, M. C., Lin, J., Pop, M., and
Salzberg, S. (2009). Searching for snps with cloud
computing. Genome Biology, 10(11):R134+.
Lenk, A., Klems, M., Nimis, J., Tai, S., and Sandholm,
T. (2009). What’s inside the cloud? an architectural
map of the cloud landscape. In Proceedings of the
2009 ICSE Workshop on Software Engineering Chal-
lenges of Cloud Computing, CLOUD ’09, pages 23–
31, Washington, DC, USA. IEEE Computer Society.
Maymounkov, P. and Mazieres, D. (2002). Kademlia: A
peer-to-peer information system based on the xor met-
ric. In Proceedings of the 1st International Workshop
on Peer-to Peer Systems, IPTPS ’02.
Mulder, N. J., Apweiler, R., Attwood, T. K., Bairoch, A.,
Bateman, A., Binns, D., Bork, P., Buillard, V., Cerutti,
L., Copley, R. R., Courcelle, E., Das, U., Daugh-
erty, L., Dibley, M., Finn, R. D., Fleischmann, W.,
Gough, J., Haft, D. H., Hulo, N., Hunter, S., Kahn, D.,
Kanapin, A., Kejariwal, A., Labarga, A., Langendijk-
Genevaux, P. S., Lonsdale, D., Lopez, R., Letunic, I.,
Madera, M., Maslen, J., McAnulla, C., McDowall, J.,
Mistry, J., Mitchell, A., Nikolskaya, A. N., Orchard,
S. E., Orengo, C. A., Petryszak, R., Selengut, J. D.,
Sigrist, C. J. A., Thomas, P. D., Valentin, F., Wil-
son, D., Wu, C. H., and Yeats, C. (2007). New de-
velopments in the interpro database. Nucleic Acids
Research, 35(Database-Issue):224–228.
Ogasawara, E., de Oliveira, D., Chirigati, F., Barbosa, C. E.,
Elias, R., Braganholo, V., Coutinho, A., and Mattoso,
M. (2009). Exploring many task computing in sci-
entific workflows. In Proceedings of the 2nd Work-
shop on Many-Task Computing on Grids and Super-
computers, MTAGS ’09, pages 2:1–2:10, New York,
NY, USA. ACM.
Pan, Q., Lee, O. S. L. J., Frey, B. J., and Blencowe,
B. J. (2008). Deep surveying of alternative
splicing complexity in the human transcriptome
by high-throughput sequencing. Nature Genetics,
40(12):1413–1415.
Qiu, X., Ekanayake, J., Beason, S., Gunarathne, T., Fox, G.,
Barga, R., and Gannon, D. (2009). Cloud technologies
for bioinformatics applications. In Proceedings of the
2nd Workshop on Many-Task Computing on Grids and
Supercomputers, MTAGS ’09, pages 6:1–6:10, New
York, NY, USA. ACM.
Richardson, L. and Ruby, S. (2007). Restful Web Services.
O’Reilly Media, Inc., 1 edition.
Schatz, M. C. (2009). Cloudburst: highly sensitive read
mapping with mapreduce. BMC Bioinformatics,
25(11):1363–1369.
Sciences, . L. (2010). 454 Life Sciences.
http://www.454.com.
Stoica, I., Morris, R., Liben-Nowell, D., Karger, D. R.,
Kaashoek, M. F., Dabek, F., and Balakrishnan, H.
(2003). Chord: a scalable peer-to-peer lookup proto-
col for internet applications. IEEE/ACM Trans. Netw.,
11(1):17–32.
Sultan, M., Schulz, M. H., Richard, H., Magen, A., Klin-
genhoff, A., Scherf, M., Seifert, M., Borodina, T.,
Soldatov, A., Schmidt, D. P. D., O’Keeffe, S., Haas,
S., Vingron, M., Lehrach, H., and Yaspo, M. L.
(2008). A global view of gene activity and alternative
splicing by deep sequencing of the human transcrip-
tome. Science, 321(5891):956–960.
Wang, J., Crawl, D., and Altintas, I. (2009). Kepler
+ hadoop: a general architecture facilitating data-
intensive applications in scientific workflow systems.
In Proceedings of the 4th Workshop on Workflows in
Support of Large-Scale Science, WORKS ’09, pages
12:1–12:8, New York, NY, USA. ACM.
White, T. (2009). Hadoop: The Definitive Guide. O’Reilly,
first edition edition.
Wilkinson, M. D. and Links, M. (2002). BioMOBY: an
open source biological web services proposal. Brief-
ings in Bioinformatics, 3(4):331–341.
A CLOUD ARCHITECTURE FOR BIOINFORMATICS WORKFLOWS
483
