
DIFFERENTIAL EVOLUTION TO MULTI-OBJECTIVE PROTEIN
STRUCTURE PREDICTION
Sandra M. Venske
1,2
, Richard A. Gonc¸alves
1
and Myriam R. Delgado
2
1
Department of Computer Science, Midwestern State University of Paran´a, Guarapuava, Brazil
2
CPGEI - Graduate Program on Electrical Engineering and Computer Science
Federal Technological University of Paran´a, Curitiba, Brazil
Keywords:
Protein structure prediction, Multi-objective optimization, Differential evolution.
Abstract:
Protein structure prediction (PSP) is one of the most challenging problems nowadays and an important Bioin-
formatics research topic. In this paper we propose an optimization method based on differential evolution
for PSP problem. We model PSP as an optimization problem in order to minimize the potential energy us-
ing ab initio approach. This problem is handled here as multi-objective optimization, and it is solved by the
evolutionary method of Differential Evolution (DE). An innovative way of choosing the best individual of the
population is proposed in this work: the minimum distance to the empirical ideal point. The idea is to guide
the population individuals to areas of the Pareto front that correspond to a good compromise of the bonded
and non-bonded energies. The proposed approach is validated on some peptides with promising results.
1 INTRODUCTION
Proteins are composed of one or more polypeptide
chains, each one containing from several to hundreds
or even thousands amino acids, and are responsible
for many different biological functions.
In order to understand the functions of proteins at
a molecular level, it is often necessary to determine
their three dimensional structure. Indeed, this infor-
mation is important to design new drugs capable of
combating diseases (Cohen and Kelly, 2003). When
a protein is in its folded state, its free energy confor-
mation is the lowest one.
In this paper we propose an optimization method
based on differentialevolution (DE) for Protein Struc-
ture Prediction (PSP), the MODE-P - Multi-Objective
Differential Evolution for PSP problem. We model
PSP as an optimization problem in order to minimize
the potential energy. This problem is handled here
as multi-objective optimization, to be solved by the
evolutionary method of DE. Our approach is an at-
tempt to contribute to the PSP problem by means of
an evolutive method - that uses a different way for
picking the individual that will guide the evolutionary
process.
2 BACKGROUND
DE is a stochastic, population-based search strategy
developed by Storn and Price (Storn and Price, 1997).
Summarized, DE adds the weighted difference be-
tween two population vectors (difference vectors) to
a third vector (target vector). The term differential
evolution comes from the fact that the process of
this evolutionary algorithm is based on difference be-
tween individuals in the population. In this paper we
used DE/best/1/bin variation, where (Storn and Price,
1997).
General Multi-Objective Optimization Problem
(MOP) is defined as minimizing (or maximizing) F(x)
= (f
1
(x), ..., f
k
(x)) subject to g
i
(x) ≤ 0, i = {1, ..., m},
and h
j
(x) = 0, j = {1, ..., p} x ∈ Ω. A solution mini-
mizes (or maximizes) the components of a vector F(x)
where x is a n-dimensional decision variable vector x
= (x
1
, ..., x
n
) from some universe Ω.
In this work we use CHARMM (v.27) force field
calculated as a function of terms for internal (bonded)
and external (interaction or non-bonded) contribu-
tions. They represent the two objective functions to
be separately minimized in the evolutionary process.
The concept of empirical ideal point used in this
work to select the best individual in DE is: let z
∗
=
(z
∗
1
, z
∗
2
, ..., z
∗
i
), where z
∗
i
∈ Z is such that f
i
(x
∗
i
) =
minf
i
(x), x ∈ A∪B and A represents the solutions int
295
M. Venske S., A. Gonçalves R. and R. Delgado M..
DIFFERENTIAL EVOLUTION TO MULTI-OBJECTIVE PROTEIN STRUCTURE PREDICTION.
DOI: 10.5220/0003767002950298
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2012), pages 295-298
ISBN: 978-989-8425-90-4
Copyright
c
2012 SCITEPRESS (Science and Technology Publications, Lda.)

the current population and B represents the solutions
in non-dominated archive. Z contains all possible val-
ues that can be assumed by F(x). In this work, the
decision maker choose the final solution based on the
empirical ideal point.
Our approach has its contribution relative to other
works ((Cutello et al., 2006), (Tudela and Lopera,
2009), (Becerra et al., 2010)) in the use and test of
DE with an innovative way of choosing the best in-
dividual used during differential mutation based on
the empirical ideal point. Also, instead of using the
knee concept (Coello et al., 2007), like most of the
above approaches does, our decision maker also uses
the empirical ideal point concept.
3 MODE-P
In the current work, in order to represent the candi-
date solutions, we adopt a model based on off-lattice
and an internal coordinates representation - the tor-
sion angles - with backbone and sidechain torsion an-
gles to model proteins. Each residue type has a pre-
established number of torsion angles in order to reach
a conformation of a protein. The backbone of each
residue is represented by 3 dihedral angles: φ, ψ, ω.
The sidechains are represented by χ
i
angles.
In order to reduce the search space, we use the
restricted range of angles showed in Sun et al. (1997).
The secondary structure constraints for peptides were
predicted here using Pollastri et al. (2002).
Our proposed approach is called MODE-P (Multi-
Objective Differential Evolution for PSP problem),
follows the basic scheme of DE with some modifica-
tions to deal with the bi-objective PSP problem. The
process involves the identification of non-dominated
solutions, their storage and further inclusion into the
population. In order to store the non-dominated so-
lutions the same storage procedure applied in Pareto
Archived Evolution Strategy (PAES) is used. The
whole procedure of MODE-P is presented by the fol-
lowing algorithm.
Reset the generation counter, g = 0;
Initialize the control parameters, F and CR;
Create and initialize the population, pop(0),
of ns individuals accordingly to the
secondary structure constraints regions;
Evaluate pop(0);
Create nondominated archive, ND_archive
for g = 1 to MAX_GEN Do
for each individual, x_i(g) belonging to
pop(g) do
Select the best individual;
Randomly select two distinct individuals;
Create the trial vector, u_i(g) by
applying the mutation operator;
Create an offspring, x’_i(g), by
applying the crossover operator;
Evaluate (x’_i(g);
if (x_i(g) dominates x’_i(g))
discard x’_i(g)
else
if (x’_i(g) dominates x_i(g))
Update ND_archive;
Replace x_i(g) by x’_i(g) in g
else
if (x_i(g) and x’_i(g) are nondominated)
and
(x’_i(g) is nondominated by ND_archive)
Update the ND_archive;
Replace x_i(g) by x’_i(g) in g
end for
end for
For each parent individual, the routines of
DE/best/1/bin are executed.
The evaluation phase calculates potential energy
from the set of angles using TINKER Molecular
Modelling Package to compute the bond and non-
bond energy values accordingly to the CHARMM
force field model.
The choice of the best individual is based on the
solution that has the shortest euclidean distance from
the empirical ideal point of the current generation.
The empirical ideal point is composed by the cur-
rent minimum values found separately for bond en-
ergy and nonbond energy at that exact moment in the
evolution. This way for choosing the best individual
is a contribution of this work.
At the end of the evolutionary process, the archive
with nondominated solutions is returned to the deci-
sion maker. In this work, the decision maker choose
the final solution based on the empirical ideal point.
The individual who has shortest euclidean distance
from the empirical ideal point is returned to user.
4 EXPERIMENTS AND RESULTS
This section reports the results obtained for 30 inde-
pendent runs (with different seeds for each run) of
MODE-P algorithm. The population size is 400 chro-
mosomes and the number of generations is 350. The
DE parameters, CR and F, are set as equal to 0.7 and
0.2, respectively. In order to assess how similar is
the predicted conformation to the native structure, the
RMSD (Root Mean Square Deviation) metric is used
(Tramontano, 2006).
MODE-P was applied to Met-Enkephalin pep-
tide (1PLW) and two others protein sequences from
PDB (Protein Data Bank): Crambin (1CRN) and
Disulphide-stabilized mini protein A domain (1ZDD).
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
296
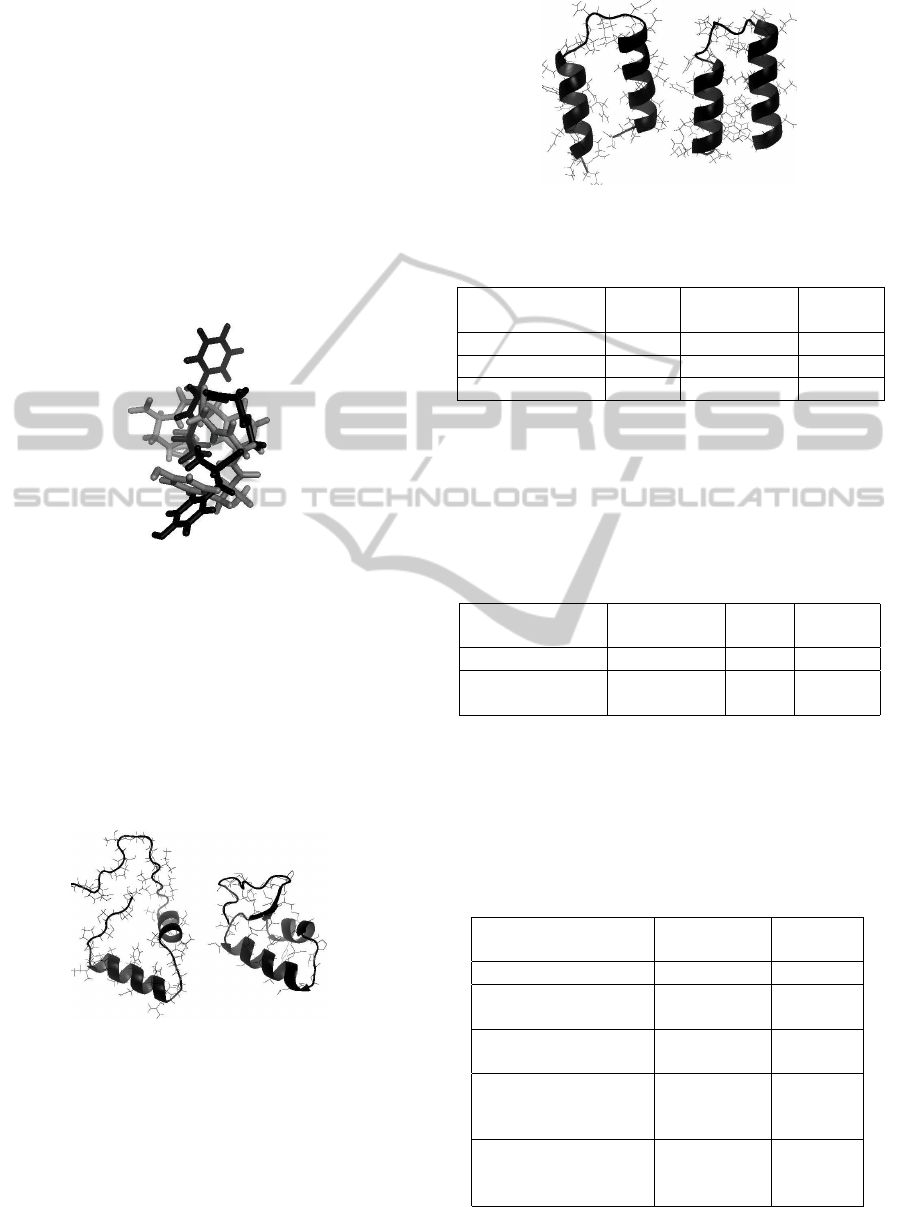
Table 1 summarizes the results for each protein.
MODE-P decision maker is applied to the archive
with nondominated solutions of the best run. We as-
sume that the best run is the one that returned the in-
dividual with the lowest energy among all executions.
Met-Enkephalin is a polipeptide with 5 amino
acids used as classical test for algorithms designed for
PSP problem. Decision maker found the solution with
energy value of -33.11 kcal mol
−1
that matches the
crystal structure of 1PLW obtained from PDB with
RMSD
all−atoms
= 3.144
˚
A e RMSD
C
α
= 1.814
˚
A. Fig-
ure 1 shows the comparison between predicted con-
formation choosen by MODE-P decision maker and
1PLW.
Figure 1: Comparison between predicted conformation
(black) and 1PLW conformation for Met-enkephalin pep-
tide. Figure generated by PyMOL.
1CRN is a 46-residue protein with two α-helix
an a pair of β-strands. MODE-P decision maker
found the solution with energy value of 408.53 kcal
mol
−1
that matches the crystal structure from PDB
with RMSD
all−atoms
= 5.590
˚
A e RMSD
C
α
= 5.559
˚
A. Figure 2 shows the comparison between predicted
conformation choosen by MODE-P decision maker
and the crystal structure of 1CRN.
Figure 2: Comparison between predicted conformation
(left) and 1CRN protein. Figure generated by PyMOL.
1ZDD is a two-helix peptide of 34 residues. De-
cision maker found the solution with energy value of
-1050.85 kcal mol
−1
that matches the crystal structure
from PDB with RMSD
all−atoms
= 6.213
˚
A e RMSD
C
α
= 3.846
˚
A. Figure 3 shows the comparison between
predicted conformation choosen by MODE-P deci-
sion maker and the crystal structure of 1ZDD.
Figure 3: Comparison between predicted conformation
(left) and 1ZDD protein. Figure generated by PyMOL.
Table 1: Results obtained by MODE-P for peptides.
Protein Amino
acids
Energy (kcal
mol
−1
)
RMSD
C
α
(
˚
A)
Met-Enkephalin 5 -33.11 1.814
1CRN 46 408.53 5.559
1ZDD 34 -1050.85 3.846
We compared the results of MODE-P algorithm
with other approaches in the literature.
Table 2 shows the results achieved by our pro-
posed approach (MODE-P) when compared with the
ones provides by Cutello et al. (2006) for Met-
enkephalin peptide.
Table 2: Results for Met-Enkephalin peptide.
Algorithm Energy (kcal
mol
−1
)
RMSD
(
˚
A)
RMSD
C
α
(
˚
A)
MODE-P -33.11 3.144 1.814
I-PAES (Cutello
et al., 2006)
-20,56 3.605 1.740
Table 3 reports the comparison of MODE-P ver-
sus other approaches for 1CRN and Table 4 compares
MODE-P with others two approaches for 1ZDD. The
RMSD
C
α
values do not apprear in these tables be-
cause such measures have been ommited in the con-
sidered literature.
Table 3: Results for 1CRN protein.
Algorithm Energy (kcal
mol
−1
)
RMSD
C
α
(
˚
A)
MODE-P 408.53 5.559
I-PAES (Cutello et al.,
2006)
701.25 4.43
Dandekar and Argos
(1996)
– 5.4
NSGA2 (with high-
level operators)
(Cutello et al., 2006)
– 6.447
NSGA2 (with low-
level operators)
(Cutello et al., 2006)
– 10.34
The results show that MODE-P is competi-
DIFFERENTIAL EVOLUTION TO MULTI-OBJECTIVE PROTEIN STRUCTURE PREDICTION
297

tive when compared with the literature. For Met-
enkephalin e 1CRN, MODE-P proved to be a good
optimizer considering the potential energy values. Its
values are smaller than those in the literature, asso-
ciate with good RMSD values. In particular case
of Met-enkephalin, for instance, potential energy and
RMSD
all−atoms
values obtained by MODE-P are bet-
ter than the comparison approach. For 1ZDD the val-
ues in terms of energy and RMSD were competitive
to others approaches.
Table 4: Results for 1ZDD protein.
Algorithm Energy (kcal
mol
−1
)
RMSD
C
α
(
˚
A)
MODE-P -1050.85 3.846
I-PAES (Cutello et al.,
2006)
-1052.09 2.27
GA (Dorn et al., 2011) -983.27 3.92
5 CONCLUSIONS AND FUTURE
WORKS
This paper has presented a multi-objective evolution-
ary algorithm for PSP problem with ab initio ap-
proach. The evaluation of the conformation of a pro-
tein is estimated using energy values of local and non-
local interactions in order to compose the potential
energy.
The results obtained suggest that MODE-P can
predict small proteins structures with competitive val-
ues compared with other works in literature. The in-
novative way for choosing the best individual in a
multi-objective differential evolution proved to be a
good option to be used during the evolutionary pro-
cess.
As future work we intend to expand MODE-P to
deal with medium size proteins and investigate alter-
native methods for decision maker.
ACKNOWLEDGEMENTS
The authors acknowledge the CNPq grant 307735/
2008-7 and Fundac¸˜ao Arauc´aria project 400/0910705
for the partial financial support.
REFERENCES
Becerra, D., Sandoval, A., Restrepo-Montoya, D., and
Ni˜no, L. F. (2010). A parallel multi-objective ab initio
approach for protein structure prediction. In Park, T.,
Tsui, S. K.-W., Chen, L., Ng, M. K., Wong, L., and Hu,
X., editors, BIBM, pages 137–141. IEEE Computer So-
ciety.
Coello, C. C., Lamont, G., and van Veldhuizen, D.
(2007). Evolutionary Algorithms for Solving Multi-
Objective Problems. Genetic and Evolutionary Compu-
tation. Springer, Berlin, Heidelberg, 2nd edition.
Cohen, F. E. and Kelly, J. W. (2003). Therapeutic
approaches to protein-misfolding diseases. Nature,
426:905–909.
Cutello, V., Narzisi, G., and Nicosia, G. (2006). A multi-
objective evolutionary approach to the protein structure
prediction problem. J R Soc Interface, 3(6):139–51.
Dandekar, T. and Argos, P. (1996). Identifying the tertiary
fold of small proteins with different topologies from se-
quence and secondary structure using the genetic algo-
rithm and extended criteria specific for strand regions.
Journal of Molecular Biology, 256(3):645–660.
Dorn, M., Buriol, L. S., and Lamb, L. C. (2011). A hybrid
genetic algorithm for the 3-d protein structure prediction
problem using a path-relinking strategy. In Evolution-
ary Computation (CEC), 2011 IEEE Congress on, pages
2709 –2716.
Pollastri, G., Przybylski, D., Rost, B., and Baldi, P. (2002).
Improving the prediction of protein secondary structure
in three and eight classes using recurrent neural networks
and profiles. Proteins: Structure, Function, and Bioin-
formatics, 47:228–235.
Storn, R. and Price, K. (1997). Differential evolution
– a simple and efficient heuristic for global optimiza-
tion over continuous spaces. J. of Global Optimization,
11(4):341–359.
Sun, Z. R., Rao, X. Q., Peng, L. W., and Xu, D. (1997). Pre-
diction of protein supersecondary structures based on the
artificial neural network method. Protein Engineering,
10(7).
Tramontano, A. (2006). Protein Structure Prediction: Con-
cepts and Applications. John Wiley and Sons.
Tudela, J. C. C. and Lopera, J. O. (2009). Parallel protein
structure prediction by multiobjective optimization. In
Proceedings of the 2009 17th Euromicro International
Conference on Parallel, Distributed and Network-based
Processing, pages 268–275, Washington, DC, USA.
IEEE Computer Society.
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
298
