
BAYESIAN NETWORK ANALYSIS OF RELATIONSHIPS BETWEEN
NUCLEOSOME DYNAMICS AND TRANSCRIPTIONAL
REGULATORY FACTORS
Bich Hai Ho, Ngoc Tu Le and Tu Bao Ho
Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, 923-1292 Ishikawa, Japan
Keywords:
Nucleosome dynamics, Bayesian network, Post-translational histone modification, Transcriptional regulator.
Abstract:
Intergenic regions are unstable, owing to trans-regulatory factors that regulate chromatin structure. Nucleo-
some organization at promoter has been shown to exhibit distinct patterns corresponding to the level of gene
expression. Post-translational modifications (PTMs) of histone proteins and transcriptional regulators, includ-
ing chromatin remodeling complexes (CRCs), general transcription factors (GTFs), and RNA polymerase II
(PolII), are presumably related to the establishment of such nucleosome dynamics. However, their concrete
relationships, especially in gene regulation, remain elusive. We, therefore, sought to understand the functional
linkages among these factors and nucleosome dynamics by deriving a Bayesian network (BN)-based model
representing their interactions. Based on the recovered network, learnt from 8 PTMs and 15 transcriptional
regulators at 4034 S.cerevisiae promoters, we speculate that nucleosome organization at promoter is intention-
ally volatile in various regulatory pathways. Notably, interactions of CRCs/GTFs and H3 histone methylation
were inferred to co-function with nucleosome dynamics in gene repression and pre-initiation complex (PIC)
formation. Our results affirm the hypothesis that extrinsic factors take part in regulating nucleosome dynamics.
More thorough investigation can be made by adding more factors and using our proposed method.
1 INTRODUCTION
Eukaryotic genomes are packaged inside cell nucleus
under chromatin structure like a bead-on-string fiber
of nucleosomes. As a fundamental unit, nucleosome
contains a core of octamer histone proteins wrapped
around by 147bp of DNA (Luger et al., 1997). More
than DNA packaging, chromatin involves in various
cellular processes such as transcription, DNA replica-
tion, etc., by occluding the access of biological ma-
chineries to cis-regulatory elements and/or modify-
ing related epigenetic information. To overcome the
obstacle imposed by chromatin, cells have developed
complicated pathways (Li et al., 2007), in which nu-
cleosome must be dislocated from chromatin to pro-
vide access to the underlying DNA sequences. While
positioning are strongly influenced by intrinsic DNA
sequence preference (Kaplan et al., 2010), the rear-
rangement can be flexibly modulated by extrinsic fac-
tors, e.g., DNA-binding factors and CRCs (Wan et al.,
2009). They help to maintain the periodicity, hence
the corresponding transcription activities, by directly
altering nucleosome organization in various manners
(Venters and Pugh, 2009). PTMs were shown in vari-
ous works to be related to nucleosome spatial organi-
zation (Cui et al., 2010). These two factors interact in
that PTMs serve as their targeting marks, and in turn,
the locations of PTMs is modulated by those regula-
tory proteins. Such series of highly regulated interac-
tions may necessarily be characterized by a network
featuring variable correlations. Since the data in use
here are all related to transcriptional activities, we ref-
erenced to the common resulting effects on gene ex-
pression to infer possible functional linkages.
Taken together, we speculated that in complex in-
teraction network, the nucleosome dynamics at pro-
moter may play an intermediate role, i.e. affecting as
well as being affected by other factors, in regulatory
pathways, assuming that there are two classes of pro-
moters, unstable with periodic nucleosome arrange-
ments, and stable without. We employed Bayesian
network (BN), a class of probabilistic graphical mod-
els that can capture not only co-occurrence pattern
but also interaction/dependency among variables, for
interaction modeling. BN has been used to recon-
struct many kinds of cellular networks, such as gene
regulatory networks and protein interaction networks
(Friedman et al., 2000). Compared with previous
299
Ho B., Le N. and Ho T..
BAYESIAN NETWORK ANALYSIS OF RELATIONSHIPS BETWEEN NUCLEOSOME DYNAMICS AND TRANSCRIPTIONAL REGULATORY
FACTORS.
DOI: 10.5220/0003773502990302
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2012), pages 299-302
ISBN: 978-989-8425-90-4
Copyright
c
2012 SCITEPRESS (Science and Technology Publications, Lda.)

findings, we showed that the built network can re-
cover known relationships as well as functional link-
ages consistent with knowledge about various DNA-
mediated processes.
2 METHOD
2.1 Datasets
The experimental data of 24 features Saccharomyces
cerevisiae were gathered as follows: Two classes
of 4043 yeast promoters, including 1355 unsta-
ble/dynamic (no periodicity and high expression) and
2688 stable (explicit periodicity and low expression)
were obtained from (Wan et al., 2009); 8 PTMs (from
−1000bp to TSSs) were obtained from (Pokholok et
al., 2005); 15 transcriptional regulators including 6
GTFs, 1 PolII component, and 8 CRCs from (Ven-
ters and Pugh, 2009). Data was discretized using
proportional k-interval discretization (PIKD) (Yu et
al., 2008) with intervals of [33%, 66%], [20%, 80%],
[40%, 60%], and determined by K-means clustering
(k = 3). As a result, [20%,80%] proportional 3-
interval scheme gave us the most reasonable network
(data not shown).
2.2 Bayesian Networks
2.2.1 Definition
A Bayesian network for a set of variables X =
{X
1
,X
2
,... , X
n
} is a probabilistic model consisting of
two components (Heckerman, et al., 1995):
• A network structure S, which is a directed acyclic
graph, representing conditional (in)dependence
relationships among variables in X
• A set P of local probability distributions associ-
ated with each variable.
Because the main target of our work is to uncover
the relationships among the PTMs so we are inter-
ested in the problem of learning BN structure. We
employed the score-based search method (Jensen and
Nielsen, 2007) to learn a BN structure representing
relationships among PTMs. To score a candidate net-
work, we used a Bayesian scoring metric, namely
BDe (Bayesian metric with Dirichlet prior and equiv-
alence) (Heckerman, et al., 1995).
2.2.2 Bootstrapping and Selection of Cut-off
Threshold
The search-and-score method generates a different
network on each run, and only one with highest score
is output. Hence, we employed the bootstrapping
method (Friedman et al., 2000) to estimate the con-
fidence level for each edge in the resulting network.
Then following hybrid method was proposed to de-
rive a suitable threshold value for confidence level of
each edge in the resulting network:
1. Divide data into n datasets using n-fold cross-
validation, t times.
2. At each time, n bootstrapped consensus BNs
N
1
.. . N
n
are output, using a fixed τ. Then, n cross-
validated networks are combined into one by in-
cluding edges agreed by θ graphs. Thus, with
each pair of (τ and θ), one final network N is
learnt.
3. To measure the goodness of the learnt network,
accuracy (acc) and coverage (cov) are used and
plotted as receiver operating characteristic (ROC)
curve. Co-ordinates of each point in the curve is
the average of t times (with standard deviation).
The chosen network has τ and θ that generate the
largest area under the curve (AUC).
acc
i
=
♯(N
i
∩ N)
♯N
i
,cov
i
=
♯(N
i
∩ N)
♯N
,i = 1, .. ., n
(1)
where the numerator is the number of overlapping
edges of each network with the common one; the
denominator is the number of edges in each net-
work and the common one, respectively.
In our experiment, we split data into 5 datasets (ac-
cording to 5-fold cross-validation) for t = 50 times;
on each, we bootstrapped 100 times (m= 100), which
totally resulted in 25, 000 input datasets for learning.
Each edge in the consensus network has a related con-
fidence score, measured by the number of times it ap-
pears in 100 bootstrapped ones. Threshold τ was cho-
sen in the range of [0.5, 0.8] with the step of 0.05. The
combined network consisted of overlapping edges by
θ = {2, 3,4} cross-validated ones. We then chose pa-
rameters θ = 3 and τ = 0.65 to produce a network
with 24 features as its nodes and 36 edges represent-
ing the functional linkages among PTMs, transcrip-
tional regulators, and nucleosome dynamics (Fig. 1).
3 RESULTS AND DISCUSSION
3.1 Network Recovers Reliable
Functional Linkages
Comparing our network (Fig. 1) with previous find-
ings, we found a remarkable consistency with original
results reported in (Venters and Pugh, 2009). CRCs
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
300
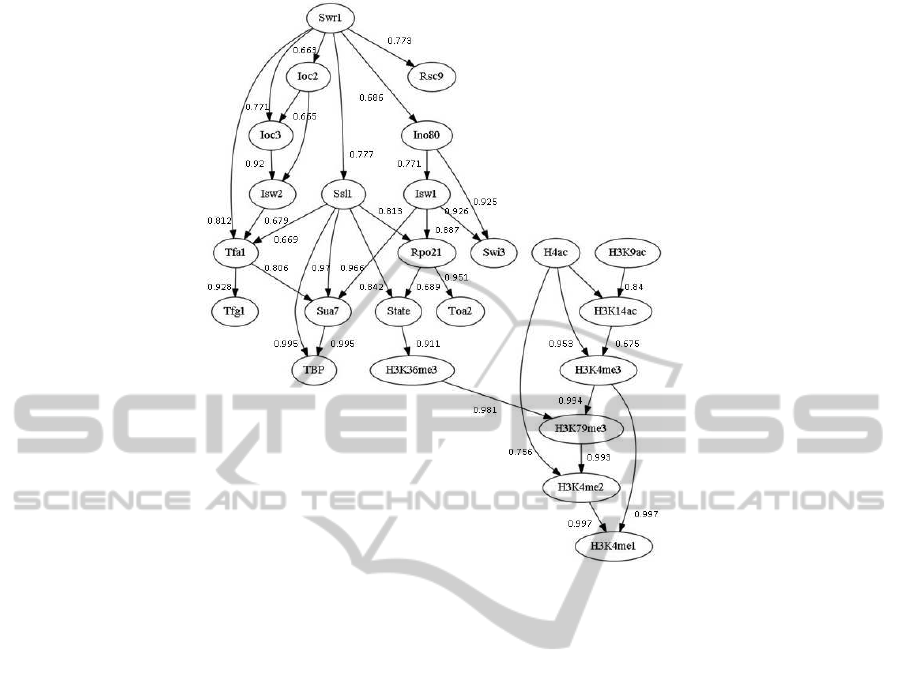
Figure 1: PTMs, TFs, and nucleosome dynamics network with confidence level (in range [0,1]). Note that the edge direction-
ality does not show definite causality, due to the nature of the learning algorithm; thus in most cases, we inferred correlations
or interactions among network nodes.
with subunits such as Swr1, Ioc2, Ioc3, Ino80, etc.,
appear in the upper part of the network, showing
causal relationships towards GTFs, which generally
reflects the role of CRCs as an ATP-dependent nucle-
osome translocator giving way to GTFs accessing the
buried DNA. From the network topology, it is clear
that nucleosomedynamics bridges two groups of tran-
scriptional regulators and PTMs, for which, unfortu-
nately, we have yet to find direct evidence. Nodes
of high out degree (number of edges pointing away
from node), such as Swr1 (out degree of 6) and Ssl1
(out degree of 5), may play a central role; Swr1, (sub-
unit of SWR-C), is implicated in the deposition of
histone variant H2A.Z in promoter, which provides
a molecular mechanism to regulate transcription and
DNA repair (Luksend et al., 2010). Especially, to-
gether with NuA4, an essential histone acetyltrans-
ferase, they function in NuA4/SWR-C/H2A.Z pathway
to regulate chromosome stability.
3.2 Nucleosome Dynamics in
Regulatory Processes
3.2.1 PIC Formation by GTFs and PolII Affects
Nucleosome Dynamics
It is agreed that GTFs assemble at promoter to form
pre-initiation complex (PIC). The interactions among
them have been well investigated, and the related sub-
graph (TFIIH(Ssl1) → TFIIE(Tfa1) → THIIE(Tfg1))
→ TFIIB(Sua7) → TBP) → PolII (Rpo21) → State
in our network is consistent with previous findings
(Samorodnitsky and Pugh, 2010). Especially, these
factors were considered in (Samorodnitsky and Pugh,
2010), a modeling work (PathCom) towards the or-
dering of PIC assembly/disassembly at the genes of
yeast. The proposed assembly order was recruit-
ment of TBP, TFIIB first, then PolII and other GTFs
(TFIIB, TFIIE, TFIIF, TFIIH in order); disassembly
goes backwards. Interpretation from our network that
the binding of factor at child node correlates with that
of parent node in an order manner, i.e., the biding
of parent affects that of child, not vice versa, is rea-
sonably consistent with this model. Hence, from our
network, nucleosome organization (node State) may
BAYESIAN NETWORK ANALYSIS OF RELATIONSHIPS BETWEEN NUCLEOSOME DYNAMICS AND
TRANSCRIPTIONAL REGULATORY FACTORS
301

inferably alter along with PIC forming (most impor-
tantly, PolII), which well matches with the known hy-
pothesis that nucleosome dynamics facilitates access
to GTFs (Morse, 2007) and CRCs.
3.2.2 Limited Gene Expression by CRCs is
Related to Periodic Nucleosome
Organization at Promoter
We analyzed the chain of SWR-C(Swr1) →
INO80(Ino80) → ISWIa/b(Isw1) → PolII(Rpo021)
→ State to understand the mechanism of gene repres-
sion, which is observable in our data as the periodic
nucleosome organization of limitedly expressed
genes. As pointed out in (Lindstrom et al., 2006),
Isw1 shows parallel functions with NuA4 and Swr1
complexes in repressing genes. ISWa/b plays a role
in repressing transcription (Pinskaya et al., 2009).
Hence, the interactions of Swr1, NuA4, Isw1, and
Rpo21 at promoter can reasonably reason the gene
repressing directionality. In our network, however,
INO80 stands as a bridge between Swr1 and Isw1,
which is explainable because INO80 is essential in
H2A.Z correct deposition (Lindstrom et al., 2011)
and may catalyze the removal of unacetylated H2A.Z.
We, therefore, speculated that the presence of NuA4
as histone acetyltransferase and INO80 as remover
of unacetylated H2A.Z at promoter in repressing
pathway defines the low level of gene expression;
and that in the pathway, INO80 help establishing the
periodicity of nucleosome organization.
4 CONCLUSIONS
We present here the reconstruction of interaction net-
work among various PTMs and transcriptional regu-
lators, with focus on their relationships with the dy-
namic nucleosome organization at promoter. Having
a large number of relationships correctly recovered,
the network features the regulatory processes that
show their presence by changing nucleosome orga-
nization at promoter, e.g., gene repression, postinitia-
tion regulation and PIC assembly/disassembly. Also,
we provide evidences for the hypothesis that nucleo-
some dynamics at promoter is regulated by extrinsic
factors, such as CRCs and GTFs. With these results,
the reliability of our method is proved, in addition to
the proposed valid learning procedure; hence, it can
be used to build networks of other factors.
REFERENCES
Cui, P., Zhanga, L., Lina, Q., Dinga, F., Xina, C., Fanga, X.,
Hua, S., and Yua, J. (2010). A novel mechanism of
epigenetic regulation: Nucleosome-space occupancy.
391(1):884–89.
Friedman, N., Linial, M., Nachman, I., and Peer, D. (2000).
Using bayesian networks to analyze expression data.
7(3-4):601–20.
Heckerman, D., Geiger, D., and Chickering, D. (1995).
Learning bayesian networks: The combination of
knowledge and statistical data. 20:197–243.
Jensen, F. and Nielsen, T. (2007). Bayesian Networks
and Decision Graphs. Springer-Verlag, New York, 2nd
edition.
Kaplan, N., Moore, I., Fondufe-Mittendorf, Y., Gossett, A.,
Tillo, D., Field, Y., Hughes, T., Lieb, J., Widom, J.,
and Segal, E. (2010). Nucleosome sequence pref-
erences influence in vivo nucleosome organization.
17:918–20.
Li, B., Carey, M., and Workman, J. (2007). The role of
chromatin during transcription. 128(4):707–19.
Lindstrom, K., Vary, J., Parthun, M., Delrow, J., and
Tsukiyama, T. (2006). Isw1 functions in parallel with
the nua4 and swr1 complexes in stress-induced gene
repression. 26(16):6117–29.
Lindstrom, K., Vary, J., Parthun, M., Delrow, J., and
Tsukiyama, T. (2011). Global regulation of h2a.z lo-
calization by the ino80 chromatin-remodeling enzyme
is essential for genome integrity. 144(2):200–13.
Luger, K., Mader, A., Richmond, R., Sargent, D., and Rich-
mond, T. (1997). Crystal structure of the nucleosome
core particle at 2.8 a resolution. 389:251–60.
Luksend, E., Ranjan, A., FitzGerald, P., Mizuguchi, G.,
Huang, Y., Wei, D., and Wu, C. (2010). Step-
wise histone replacement by swr1 requires dual ac-
tivation with histone h2a.z and canonical nucleosome.
143(5):725–36.
Morse, M. (2007). Transcription factor access to promoter
elements. 102(3):560–70.
Pinskaya, M., Nair, A., Clynes, D., Morillon, A., and Mel-
lor, J. (2009). Nucleosome remodeling and transcrip-
tional repression are distinct functions of isw1 in sac-
charomyces cerevisiae. 29(9):241930.
Pokholok, D., Harbison, C., Levine, S., Cole, M., Han-
nett, N., Lee, T., Bell, G., Walker, K., Rolfe, P.,
Herbolsheimer, E., Zeitlinger, J., Lewitter, F., Gif-
ford, D., and Young, R. (2005). Genome-wide map
of nucleosome acetylation and methylation in yeast.
122(8):517–27.
Samorodnitsky, E. and Pugh, B. (2010). Genome-wide
modeling of transcription preinitiation complex disas-
sembly mechanisms using chip-chip data.
Venters, B. and Pugh, B. (2009). A canonical promoter or-
ganization of the transcription machinery and its reg-
ulators in the saccharomyces genome. 19(3):360–71.
Wan, J., Lin, J., Zack, D., and Qian, J. (2009). Relating
periodicity of nucleosome organization and gene reg-
ulation. 25(41):1782–8.
Yu, H., S, S. Z., Zhou, B., Xue, H., and Han, J. (2008).
Inferring causal relationships among different histone
modifications and gene expression. 18(8):1314–24.
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
302
