
AN EFFICIENT PARALLEL GPU EVALUATION OF SMALL ANGLE
X-RAY SCATTERING PROFILES
Lubomir D. Antonov
1,∗
, Christian Andreetta
1,∗
and Thomas Hamelryck
1
1
The Bioinformatics Section, Department of Biology, University of Copenhagen, Copenhagen, Denmark
∗
These authors contributed equally to this work
Keywords:
SAXS, GPGPU, Protein Structure Determination.
Abstract:
The inference of protein structure from experimental data is of crucial interest in science, medicine and
biotechnology. Unfortunately, high-resolution experimental methods can not yet provide a detailed analy-
sis of the ensemble of conformations adopted under physiological conditions. Low resolution techniques are
often better suited for this task. Small angle X-ray scattering (SAXS) plays a major role in investigating im-
portant biological questions regarding the structure of multidomain proteins connected by flexible linkers or
the aggregation processes that underlie several major diseases in humans.
In silico simulations can bridge the gap between low resolution information and models derived from high-
resolution techniques. For that, it is necessary to be able to calculate the low resolution information from a
given detailed model using a so-called forward model. These calculations need to be performed many times
during a conformational search, and therefore need to be computationally efficient.
We present an efficient implementation of the forward model for SAXS experiments with full hardware utiliza-
tion of General Purpose Graphical Processor Units (GPGPUs). The proposed algorithm is orders of magnitude
faster than an efficient CPU implementation, and implements a caching procedure ready to be employed in the
partial SAXS evaluations required by in silico simulations.
1 INTRODUCTION
Proteins play a crucial role in science, medicine and
biotechnology: without them, cellular activities such
as catalysis, signaling and regulation would be impos-
sible. Protein function is determined by protein struc-
ture, which is in term determined by the amino acid
sequence (Anfinsen, 1973).
Despite encouraging improvements, determining
the ensemble of possible conformations in solution
is far from an accomplished goal. High resolution
experimental methods, notably X-ray crystallography
and Nuclear Magnetic Resonance (NMR), can only
partially provide information on such ensembles, and
encounter several limitations in fully describing the
flexibility of large systems in physiological condi-
tions (Zheng and Doniach, 2005).
Low resolution methods, on the other hand, can
more easily provide information on such ensembles.
In particular, Small Angle X-ray Scattering (SAXS)
provides information on the excess electron density of
the sample versus the surrounding environment. Re-
cently, with the advent of automated high-throughput
SAXS analysis of biomolecules (Toft et al., 2008;
Hura et al., 2009), high-throughput data acquisition
is within reach. Since SAXS data only describes the
spherical averaging of the electron density of the av-
erage conformation of the ensemble, additional infor-
mation is needed to assist structural interpretation.
Usual in silico methods propose plausible struc-
tural conformations, and calculate their associated
scattering profile by means of a forward model. Then,
the proposed and the experimental profiles are com-
pared using an error model of the experiment. For
this procedure to be successful, an efficient procedure
for both sampling protein structures and calculating
the SAXS profile is required. We focus on the latter
problem.
Early approaches for the evaluation comprise ab
initio shape determinations using spherical harmon-
ics expansions (Svergun and Stuhrmann, 1991). Here,
computational feasibility is attained at the cost of
limiting the possible shapes of the sample, for in-
stance excluding internal cavities (Koch et al., 2003).
Other modeling approaches compute and fit the scat-
tering curve from a condensation of a gas of “dummy
beads” to an experimental SAXS curve. This has been
done using conformational searches by a genetic al-
102
D. Antonov L., Andreetta C. and Hamelryck T..
AN EFFICIENT PARALLEL GPU EVALUATION OF SMALL ANGLE X-RAY SCATTERING PROFILES.
DOI: 10.5220/0003781501020108
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2012), pages 102-108
ISBN: 978-989-8425-90-4
Copyright
c
2012 SCITEPRESS (Science and Technology Publications, Lda.)

gorithm in DALAI GA (Chacon et al., 1998) or sim-
ulated annealing in DAMMIN (Svergun, 1999) and
DAMMIF (Franke and Svergun, 2009).
Higher resolution models impose further con-
straints to the proposals. In the GASBOR program
(Svergun et al., 2001), a SAXS profile is calculated
for a packed assembly of spheres placed accord-
ing to a pseudo-C
α
chain. The scattering intensity
is calculated using the Debye formula, while simu-
lated annealing is used for searching the conforma-
tional space. Other recent structure prediction meth-
ods, such as the ORNL (Tjioe and Heller, 2007) and
IMP (F
¨
orster et al., 2008) programs, utilize the SAXS
curve as an energy term in combination with other en-
ergy contributions from physical force fields. Since
these descriptors are non-probabilistic in nature, the
weights used for scaling the SAXS energy to other
conformational constraints must be chosen heuristi-
cally (Habeck et al., 2006).
In recent publications, our group developed prob-
abilistic models for the proposal of protein-like con-
formations, in full atomic detail, for both backbone
and side chains (Boomsma et al., 2008; Harder et al.,
2010). We also developed a forward model of the
scattering profile evaluation, that includes the ex-
perimental error associated with SAXS data (Stov-
gaard et al., 2010). The forward model consists of a
coarse-grained computation based on the Debye for-
mula (Debye, 1915). Our main aim is the study of
proteins consisting of multiple domains connected by
flexible linkers. Such proteins play a major role in
the regulation of gene expression, cell growth, cell
cycle, metabolic pathways, signal transduction, pro-
tein folding and transport (Levitt, 2009; Madl et al.,
2010). With this aim, a computationally efficient for-
ward model for the calculation of SAXS curves is
paramount.
We ported our original implementation of the De-
bye formula to General Purpose Graphics Process-
ing Units (GPGPUs), parallel computing engines that
can offer great advantages in terms of cost-efficiency
and low power consumption (G
¨
oddeke and Strzodka,
2011). One of the emerging standards of choice for
their programming is the Open Computing Language
(OpenCL), an open standard that provides an abstrac-
tion layer over hardware implementations of highly
parallel computational engines (Stone et al., 2010).
2 METHODS
2.1 Forward SAXS Computation
The observed data in a SAXS experiment is a one-
dimensional intensity curve, I(q), measured at dis-
cretized scattering momenta q = 4πsin(θ)/λ, with λ
the wavelength of the incoming radiation and 2θ the
scattering angle between the principal and the prob-
ing beam rays. The calculation of a theoretical SAXS
profile from a given atomic structure is based on the
well-known Debye formula (Debye, 1915):
I (q) =
M
∑
i=1
M
∑
j=1
F
i
(q)F
j
(q)
sin(q · r
i j
)
q · r
i j
(1)
where F
i
and F
j
are the scattering form factors of the
individual particles i and j, and r
i j
is the Euclidean
distance between them. The summations run over all
the M scattering particles.
2.2 Coarse-grained Protein Models
If some scatterers are fixed relative to each other, they
can be approximated by a single large scattering body
(dummy body). This approximation, more precise at
low q, fades with the progression of the scattering an-
gle up to a resolution equal to the scattering diameter
of the dummy body. We found that the amino acids
constituent to the protein chain can be approximated
by one or two large bodies (dummy atoms), and that
this approximation holds up to scattering angles nor-
mally not measured in the current experimental stan-
dards (Stovgaard et al., 2010).
In the two body model, the amino acids are indi-
vidually represented by two dummy atoms; one rep-
resenting the backbone, and the other representing
the side chain. Glycine and alanine, lacking a side
chain, are represented by one dummy atom only. The
dummy atoms are placed at the respective centers of
mass (see Figure 1). A total of 21 form factors need to
be estimated for the two body model: one for alanine,
one for glycine, one for the generic backbone and 18
for the remaining side chains.
For the one body model, the single dummy atom
is placed at the center of mass of the amino acid.
Hence, 20 form factors need to be estimated; one
for each amino acid type. For a given protein, the
one body model allows to represent the molecule in
roughly half the number of scattering bodies as com-
pared to the two body model. If the experimental data
is recorded at low resolutions only, the former is thus
clearly preferable for reasons of computational effi-
ciency.
2.3 Form Factor Descriptors
Due to the lack of publicly available high-quality
experimental data needed for the estimation of the
AN EFFICIENT PARALLEL GPU EVALUATION OF SMALL ANGLE X-RAY SCATTERING PROFILES
103
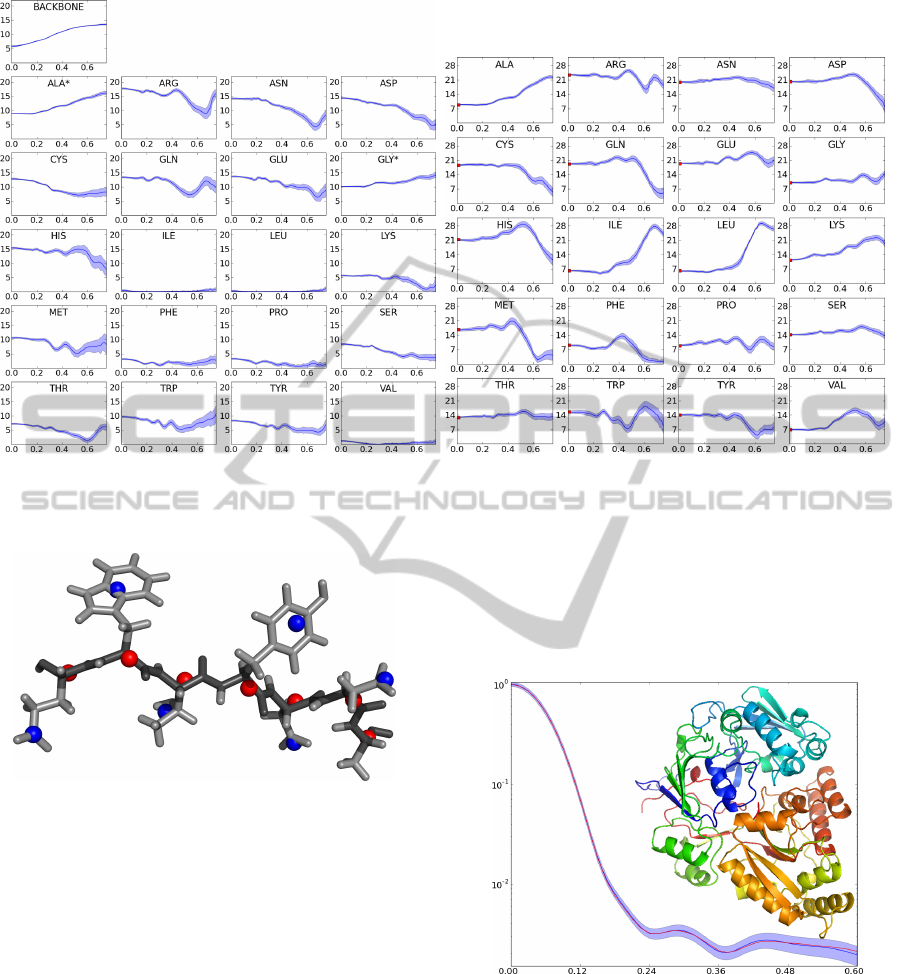
Figure 2: Form factors. Mean (dark blue curve) and standard deviations (blue areas) for the form factors (Y -axis) as a
function of q (X-axis). Left: backbone and side-chains. An asterisk indicates that this form factor describes both the backbone
and side chain atoms of the residue. Right: the single body form factors. Figure adapted from (Stovgaard et al., 2010).
Figure 1: Coarse-grained models of protein structure.
Example of a protein backbone stretch (dark gray) with side
chain atoms (light gray). The placement of the dummy
bodies for the center of mass of the backbone atoms (red
spheres) and for the side chains (blue spheres) are indicated.
Figure prepared with PyMOL (Schr
¨
odinger, 2010), adapted
from (Stovgaard et al., 2010).
form factors, artificial data curves were generated
for known high-resolution protein structures using
the state-of-the-art program CRYSOL (Svergun et al.,
1995). This program computes the theoretical scat-
tering curve from a given full-atom structure using
spherical harmonics expansions, therefore limiting its
applications at studying compact quasi-globular pro-
teins. We can however use this input to feed a learning
protocol, and make use of the Debye formula in eq. 1
to overcome structural assumptions.
Therefore, a large scale Monte Carlo simulation
has been conducted to estimate the values of the form
factors of the dummy atoms (Stovgaard et al., 2010).
The resulting profiles for these descriptors are shown
in Figure 2.
In Figure 3 we show a SAXS curve generated with
our method, and the theoretical scattering computed
by CRYSOL as a reference.
Figure 3: SAXS profile reconstruction example. Compar-
ison of the reference profiles I(q) computed by CRYSOL
from the all atom structure (blue) and by the two body
model (red). Error shade indicates the simulated “exper-
imental” error. PDB code 1JET (520 residues). Cartoon
made with PyMOL (Schr
¨
odinger, 2010).
2.4 OpenCL Programming Model
An OpenCL program contains a host program that ex-
ecutes on the CPU, and kernels that execute on the
abstracted parallel hardware. The hardware is defined
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
104
as one or more compute units, which are composed of
one or more processing elements and, in some cases,
local memory.
The host program coordinates the execution of the
kernels, and can be written in any programming lan-
guage. Kernels are written in a variant of the latest
released C language standard (C99) and are compiled
at run time to device-specific instructions. A ker-
nel describes the operations of a single work-item, or
thread, and is run simultaneously by a set of work-
items called a work-group.
The local memory of a compute unit, if present,
is shared by all work-items in a work-group and
provides an efficient communication channel among
them. It has very low latency and is usually imple-
mented with a full crossbar interface, but is limited in
size and does not retain its state between kernel exe-
cutions.
Therefore, kernels execute most efficiently when
the size of the work-group matches the size of the
compute unit on the OpenCL device and when all
work-items in a work-group follow the same execu-
tion path.
2.5 Efficient GPU Implementation
2.5.1 Parallel Page-tile SAXS Algorithm
The computation of a SAXS profile is experimentally
discretized in a set of q points, and thus naturally pro-
vides the first level of parallelization, into pages. Each
page represents the computation of the intensity curve
I(q) for a single value of q. A page can be visual-
ized as a square problem matrix of side equal to the
number of scattering particles M, with each cell repre-
senting the contribution of a single term of the Debye
formula for particular i and j.
For performance considerations and direct map-
ping to the hardware, pages are partitioned into square
tiles of side k, where k is set to the specific compute
unit size of the OpenCL device. Since each problem
matrix is symmetrical, only the tiles encompassing
the lower-left triangle and the diagonal are computed
and their value is simply duplicated for the mirror tiles
in the upper-right triangle of the matrix. The domain
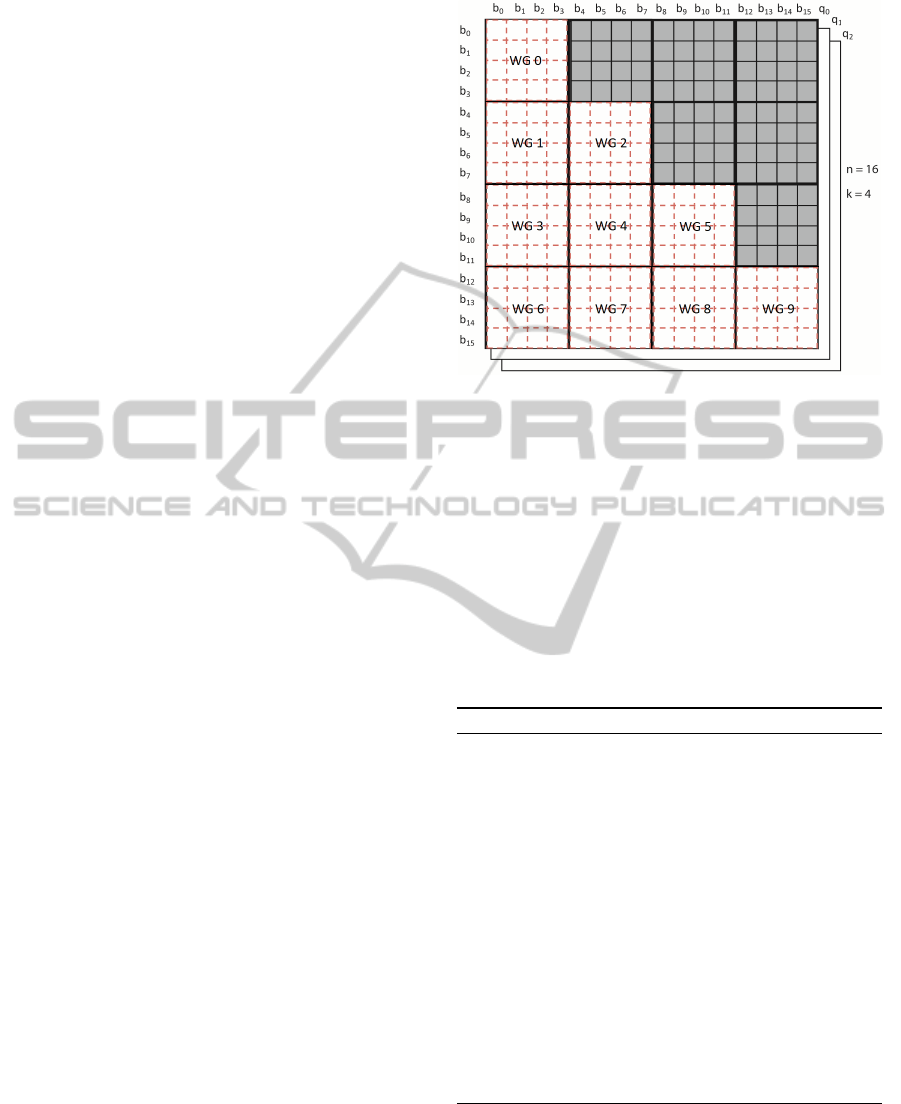
decomposition is illustrated in Figure 4 for an exam-
ple of 16 scattering particles and work-group size of
4.
GPUs suffer performance penalties when they
have to work with data that is not aligned to their na-
tive architecture, therefore the algorithm pads the data
and aligns it to the specified work-group size. The re-
sultant dummy particles participate in the Debye cal-
culations, but they are assigned a form factor of 0, so
Figure 4: Domain decomposition for the Page-Tile algo-
rithm. Work-groups operate on square tiles from the ma-
trix. Only tiles in the lower-left part and the diagonal are
evaluated.
their contribution to the intensity I(q) is null.
Algorithm 1 presents the pseudocode for the Page-
Tile SAXS algorithm. The form factors table, sup-
plied as input, is packed and organized by scattering
momentum and particle type. The scattering particles,
in addition to their position in three dimensions, have
a type in the form of an index into the form factor ta-
ble. The initial intensity curve calculation comprises
four kernels.
Algorithm 1: Page-Tile SAXS algorithm.
Input: scattering momenta, form factors table, scat-
tering particles
Output: intensity curves for the scattering momenta
/*Host program*/
Initialize the parallel algorithm
Transfer input data to GPU global memory and queue
the kernels for initial profile calculation
/*Kernels executed on the GPU*/
Map form factors to scattering particles (Kernel 1)
Compute the Debye sum term for each tile (Kernel 2)
Perform vertical tile sum reduction for each page
(Kernel 3)
Perform horizontal margin sum reduction for each
page to get the intensity curve (Kernel 4)
/*Host program*/
Retrieve the results from GPU global memory
In Kernel 1 the form factor table is mapped into
a form suited for hardware-efficient parallel access.
The form factors are organized by scattering particle,
which enables the work-groups with streaming mem-
ory access for both center coordinates and form fac-
tors.
AN EFFICIENT PARALLEL GPU EVALUATION OF SMALL ANGLE X-RAY SCATTERING PROFILES
105
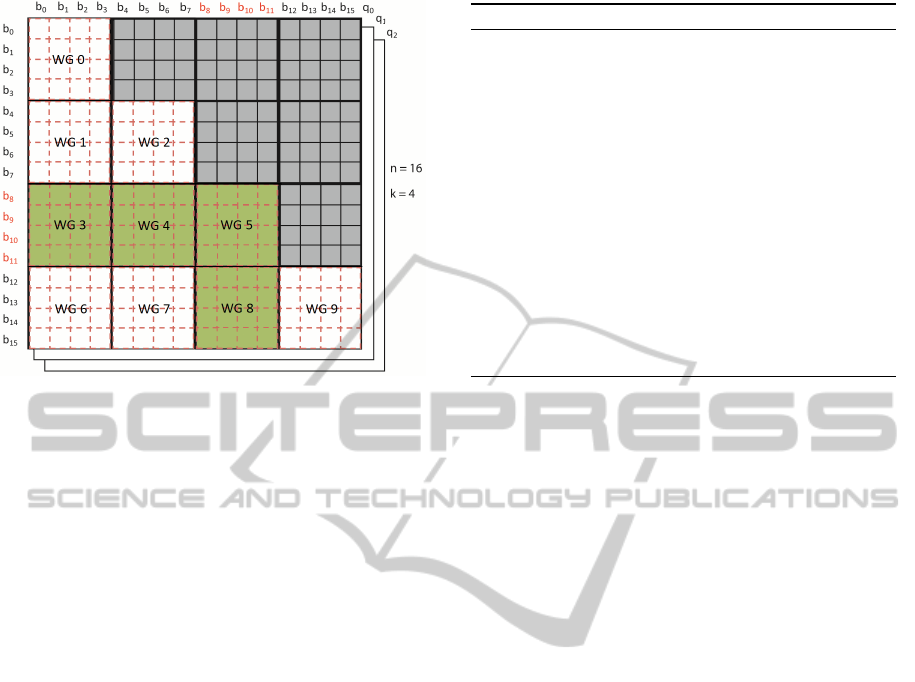
Figure 5: Problem matrix after a move. Particles b8, b9,
b10 and b11 have changed positions. Blocks WG3, WG4,
WG5 and WG8 will be recalculated.
The majority of execution time is spent in Ker-
nel 2, where the Debye sum terms for the individual
tiles are computed. The Debye formula is used for
each term, but i and j are limited to the ranges defined
by the boundaries of the tile within the global index
space. The kernel uses local memory to improve per-
formance, by pre-loading the particles and their form
factors, and by performing an in-place parallel reduc-
tion to produce the partial sum for the tile. During
the Debye calculation, 4x loop unrolling utilizes local
registers to further optimize this stage.
Kernel 3 reduces the tile partial sums, which are
stored in a global cache, to bottom margin sums that
are further reduced by Kernel 4 to yield the final in-
tensity curve.
2.5.2 Tile Recalculation
Monte Carlo Markov Chain simulations explore the
conformational space of the protein structures by ap-
plying partial modifications over an accepted pro-
posal. The average SAXS computation is therefore
a partial re-evaluation of a previously computed pro-
file, where only a subset of the bodies changed their
position.
It is therefore possible to identify the subset of
tiles that needs to be updated. Since the Page-Tile al-
gorithm caches the partial contribution of each tile to
the global summation, we can impose a partial recal-
culation of only the affected tiles (see Figure 5). This
leads to a substantial reduction in the time necessary
to derive an intensity curve after a move.
Algorithm 2 illustrates the pseudocode for tile re-
calculation. The form factor table from the initial cal-
Algorithm 2: Tile recalculation.
Input: moved scattering particles
Output: updated intensity curve for the scattering
momenta
/*Host program*/
Transfer input data to the GPU global memory and
queue the kernels involved in the profile recalculation
/*Kernels executed on the GPU*/
Compute the Debye sum term for the changed tiles
(Kernel 5)
Perform the vertical tile sum reduction for each page
(Kernel 3)
Perform horizontal margin sum reduction for each
page to get the intensity curve (Kernel 4)
/*Host program*/
Retrieve the results from the GPU global memory
culation is reused, so execution starts directly with
Kernel 5, which identifies the affected tiles and in-
vokes Kernel 2 for them. Kernel 3 and Kernel 4 per-
form the reductions as in the initial calculation.
2.5.3 Floating Point Precision
Floating point numbers can be stored and manipu-
lated with single precision (SP) or double precision
(DP). Mathematical operations on floating point num-
bers introduce errors, due to the finite precision avail-
able. Those errors tend to accumulate when a large
number of operations is performed, as is the case
with the double sum of the Debye formula. However,
the Page-Tile algorithm significantly reduces this er-
ror growth, because its successive partitioning of the
problem space results in an execution pattern resem-
bling pairwise summation (Higham, 1993). The algo-
rithm can be executed with SP or DP, paying a perfor-
mance penalty of a factor of 2 to 4 with DP.
We measured the divergence between the SP and
DP executions, and no significant differences arise be-
tween the results. Therefore, the SP implementation
is used by default.
3 RESULTS AND DISCUSSION
3.1 Computational Efficiency of the
SAXS Modeling
The Debye formula (Equation 1) leads to a com-
putational complexity of O(M
2
), with M the num-
ber of scatterers in the structure under examina-
tion. Our coarse-grained approach reduces M by rep-
resenting several atoms by one scattering body (a
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
106
dummy atom), thereby lowering the complexity to
O
(
M
/k)
2
, with k the number of scatterers (atoms)
described by a dummy body.
The precise value of k is dependent on the primary
sequence of the protein. On large datasets, the two
dummy model leads to an average k of 4.24 (with a
performance increase of k
2
' 18). The single body
model leads to k ' 7.8, allowing for a k
2
' 60 times
quicker execution.
3.2 GPGPU Implementation
The performance of the Page-Tile algorithm was mea-
sured on a system with a Core i7-920 CPU, 12GB
of DDR3 RAM and a NVIDIA GeForce GTX 560
Ti GPU with 1GB of GDDR5 RAM. The GTX
560 Ti has 8 compute units with 32 processing el-
ements each, comprising 384 processing elements,
with 32KB 32-bit registers and 48KB of local mem-
ory for each compute unit.
Performance was measured against a test protein
of over a thousand amino acids, modeled with 1888
scattering bodies in the dual dummy atom represen-
tation, and a discretization of the q space in 51 scat-
tering momenta. Protein moves were modeled by a
random mutation of 40% of the particles, to approxi-
mate the asymptotic move rate in a Monte Carlo sim-
ulation. The execution times for the model test case
are presented in Table 1.
Table 1: Execution times for SAXS curve calculation for
a protein with 1888 bodies, 51 scattering momenta and 21
form factors per momentum. Execution times from the top
are for a single processor CPU implementation, a parallel
GPU full computation, and GPU partial computation, re-
spectively. Partial computations mimic the costs in a Monte
Carlo simulation, where at each step around 40% of the pro-
posal structure is updated.
Algorithm Time (ms)
CPU SP Time 2408
GPU full calculation 9
GPU recalculation 6.484
The performance of the algorithm was also mea-
sured for protein sizes ranging from 64 to 8192 scat-
tering particles. Each protein was moved 1000 times,
in order to obtain an average of the recalculation
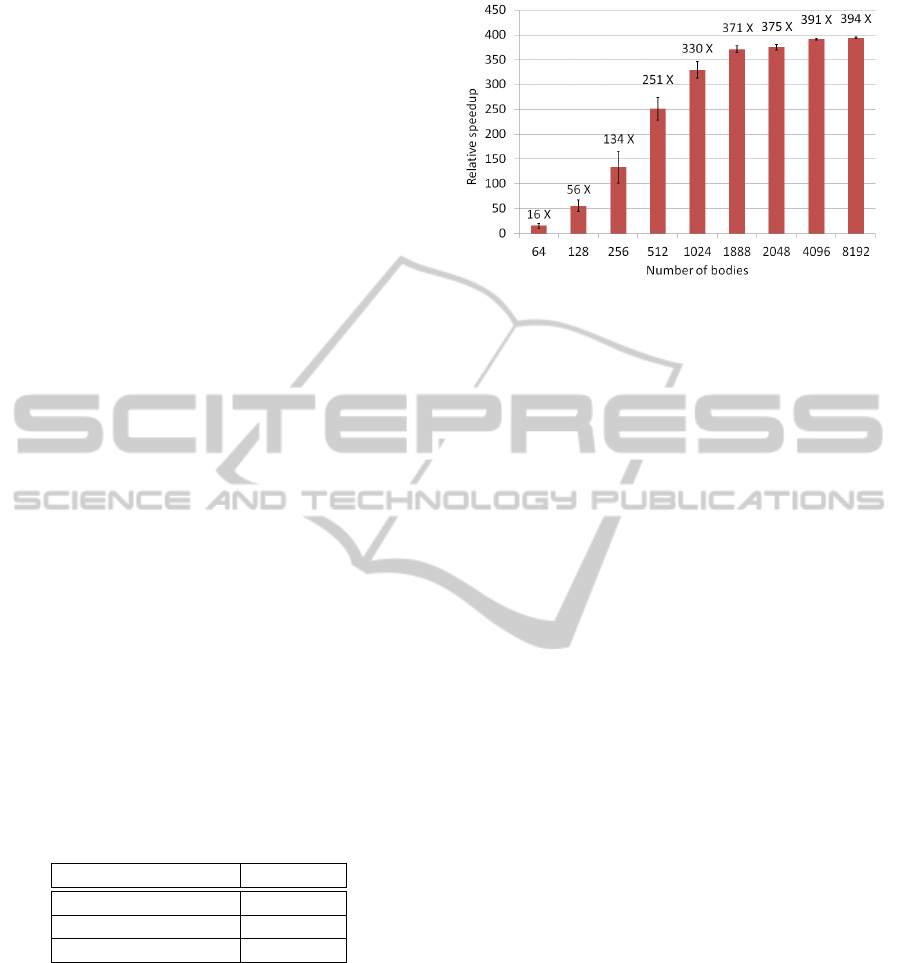
steps. Figure 6 shows the speed increase, relative to
the CPU single precision implementation, calculated
as t
cpu
/t
gpu
.
Figure 6 also illustrates the hardware utilization
of the parallel Page-Tile algorithm. The plot shows
an asymptotic behavior around problem sizes of 2000
scattering bodies. The GTX 560 Ti GPU employed in
the tests is composed of 8 compute units operating on
Figure 6: Algorithm performance for problem sizes rang-
ing from 64 to 8192 scattering bodies, including the model
test case of 1888, with error bars showing standard devia-
tion. All SAXS computations involve the summations over
51 scattering momenta. The asymptotic behavior indicates
hardware saturation at around 2000 bodies, which is the the-
oretical maximum for the GPU model used in the tests.
8 cascading work-groups, allowing for a theoretical
peak of 64 active work-groups. The work-group size
is 32, therefore the card would reach theoretical peak
processing power at 2048 bodies. Our tests show sat-
uration at the same level, thus indicating optimal use
of the hardware.
OpenCL is thread-safe and allows access to the
same device from multiple processes and threads, so
by creating multiple instances of the Page-Tile algo-
rithm, more than one calculation can be run at the
same time. This is especially relevant in the case of
problem sizes that would not lead to a full GPU sat-
uration, therefore allowing for multi-threaded Monte
Carlo simulations.
As a proof of concept, two instances of the algo-
rithm were run in parallel on the test system with the
model test problem of size 1888. The recorded time to
complete a move recalculation was 11.962ms, or 93%
of the time to complete two moves sequentially, indi-
cating that the excess processing capacity was indeed
utilized.
4 CONCLUSIONS
We have presented an efficient implementation of the
forward model for the computation of Small Angle
X-ray Scattering profiles. The application is multi-
thread safe, and benchmarks show that the General
Purpose Graphic Processor Units deliver the full the-
oretical output allowable by the hardware.
Parallelization is achieved on multiple levels by
taking advantage of the structure of the Debye prob-
lem. The first level divides the SAXS evaluation in
AN EFFICIENT PARALLEL GPU EVALUATION OF SMALL ANGLE X-RAY SCATTERING PROFILES
107

multiple independent computations according to the
binning of the scattering momentum. A nested level
then makes full use of the work-groups in the hard-
ware by splitting the inner summation of the Debye
formula in separate partial sums. The resulting pro-
gram runs orders of magnitude faster than an opti-
mized single core CPU implementation.
A caching algorithm on the inner contributions
allows for the efficient re-evaluation of SAXS pro-
files from partially updated structures, delivering even
greater benefits to applications like Markov Chain
Monte Carlo simulations of important biological and
clinical targets. An open source implementation will
be released shortly.
ACKNOWLEDGEMENTS
This research was funded by the Danish Council for
Independent Research (FTP, 274-09-0184).
SOURCE CODE AVAILABILITY
The source code of our implementation can be
found online at: http://www.phaistos.org and http://
sourceforge.net/projects/phaistos/files.
REFERENCES
Anfinsen, C. B. (1973). Principles that govern the folding
of protein chains. Science, 181(96):223–230.
Boomsma, W., Mardia, K., Taylor, C., Ferkinghoff-Borg, J.,
Krogh, A., and Hamelryck, T. (2008). A generative,
probabilistic model of local protein structure. Proc
Natl Acad Sci U S A, 105(26):8932–8937.
Chacon, P., Moran, F., Diaz, J., Pantos, E., and Andreu, J.
(1998). Low-resolution structures of proteins in so-
lution retrieved from x-ray scattering with a genetic
algorithm. Biophys J, 74:2760–2775.
Debye, P. (1915). Zerstreuung von rontgenstrahlen. Ann
Phys, 351(6):809–823.
F
¨
orster, F., Webb, B., Krukenberg, K., and Tsuruta, H.
(2008). Integration of small-angle x-ray scattering
data into structural modeling of proteins and their as-
semblies. J Mol Biol, 382:1089–1106.
Franke, D. and Svergun, D. I. (2009). Dammif, a program
for rapid ab-initio shape determination in small-angle
scattering. J Appl Crystallogr, 42(2):342–346.
G
¨
oddeke, D. and Strzodka, R. a. (2011). Cyclic reduc-
tion tridiagonal solvers on GPUs applied to mixed
precision multigrid. IEEE Transactions on Par-
allel and Distributed Systems, 22(1):22–32. doi:
10.1109/TPDS.2010.61.
Habeck, M., Rieping, W., and Nilges, M. (2006). Weighting
of experimental evidence in macromolecular struc-
ture determination. Proc Natl Acad Sci U S A,
103(6):1756–1761.
Harder, T., Boomsma, W., Paluszewski, M., Frellsen, J., Jo-
hansson, K. E., and Hamelryck, T. (2010). Beyond
rotamers: a generative, probabilistic model of side
chains in proteins. BMC Bioinformatics, 11:306.
Higham, N. J. (1993). The accuracy of floating point sum-
mation. SIAM J. Sci. Comput, 14:783–799.
Hura, G. L., Menon, A. L., Hammel, M., Rambo, R. P.,
Poole, F. L., Tsutakawa, S. E., Jenney, F. E., Classen,
S., Frankel, K. A., Hopkins, R. C., jae Yang, S., Scott,
J. W., Dillard, B. D., Adams, M. W. W., and Tainer,
J. A. (2009). Robust, high-throughput solution struc-
tural analyses by small angle x-ray scattering (saxs).
Nat Methods, 6(8):606–614.
Koch, M., Vachette, P., and Svergun, D. (2003). Small-
angle scattering: a view on the properties, structures
and structural changes of biological macromolecules
in solution. Q Rev Biophys, 36(2):147–227.
Levitt, M. (2009). Nature of the protein universe. Proc Natl
Acad Sci U S A, 106(27):11079–11084.
Madl, T., Gabel, F., and Sattler, M. (2010). NMR and small-
angle scattering-based structural analysis of protein
complexes in solution. J Struct Biol, pages 1–11.
Schr
¨
odinger, L. (2010). The PyMOL molecular graphics
system, version 1.3r1.
Stone, J. E., Gohara, D., and Shi, G. (2010). Opencl: A par-
allel programming standard for heterogeneous com-
puting systems. Computing in Science and Engineer-
ing, 12:66–73.
Stovgaard, K., Andreetta, C., Ferkinghoff-Borg, J., and
Hamelryck, T. (2010). Calculation of accurate small
angle X-ray scattering curves from coarse-grained
protein models. BMC Bioinformatics, 11:429.
Svergun, D. (1999). Restoring low resolution structure
of biological macromolecules from solution scattering
using simulated annealing. Biophys J, 76:2879–2886.
Svergun, D., Barberato, C., and Koch, M. (1995). Crysol -
a program to evaluate x-ray solution scattering of bi-
ological macromolecules from atomic coordinates. J
Appl Crystallogr, 28:768–773.
Svergun, D., Petoukhov, M., and Koch, M. (2001). Deter-
mination of domain structure of proteins from x-ray
solution scattering. Biophys J, 80(6):2946–2953.
Svergun, D. I. and Stuhrmann, H. B. (1991). New develop-
ments in direct shape determination from small-angle
scattering. 1. theory and model calculations. Acta
Crystallogr A, 47(6):736–744.
Tjioe, E. and Heller, W. (2007). Ornl sas: software for cal-
culation of small-angle scattering intensities of pro-
teins and protein complexes. J Appl Crystallogr,
40:782–785.
Toft, K., Vestergaard, B., Nielsen, S., and Snakenborg, D.
(2008). High-throughput small angle x-ray scattering
from proteins in solution using a microfluidic front-
end. Anal Chem, 80(10):3648–3654.
Zheng, W. and Doniach, S. (2005). Fold recognition aided
by constraints from small angle x-ray scattering data.
Protein Eng Des Sel, 18(5):209–219.
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
108
