
A NOVEL ANALYSIS FLOW FOR FUSED TRANSCRIPTS
DISCOVERY FROM PAIRED-END RNA-SEQ DATA
F. Abate
1
, G. Paciello
1
, A. Acquaviva
1
, E. Ficarra
1
, A. Ferrarini
2
, M. Delledonne
2
and E. Macii
1
1
Politecnico di Torino, Department of Control and Computer Engineering, Torino, Italy
2
Universit`a di Verona, Department of Biotechnology, Verona, Italy
Keywords:
Next generation sequencing, RNA-Seq data, Chimeric transcript detection, Gene fusions, Alternative splicing,
Deep sequencing analysis, Paired-end reads.
Abstract:
Chimeric phenomena have been recently recognized to play a significant role in the investigation and under-
standing of the fundamental mechanisms behind highly diffused pathologies such as tumors. In this paper we
present a new methodology for the detection of fusion transcript from Next Generation Sequencing (NGS)
data. The methodology exploits short paired-end reads coming from RNA-Seq experiments to determine a
list of fused genes and to exactly identify the fusion boundaries, so that the exact chimeric sequence can be
analysed. Both known and unknown transcripts are considered, enabling the detection of fusions involving
unannotated genes. An automated toolflow that reports a set of candidate fused genes and the associated
junctions has been implemented and applied to a publicly available data set of melanoma.
1 INTRODUCTION
Next Generation Sequencing (NGS) Technologies
have been demonstrated to play a fundamental role
in biological and genetic research fields mainly for
their capability of detecting genomic structural varia-
tions, novel genes and transcript isoforms from high
throughput data (Magalhes, 2010) (Kircher, 2010).
In particular these features are clearly recognizable
from RNA-Seq data analysis that allows a digital-
ized and sensitive estimation of gene expression lev-
els, the discover of new transcripts and also the detec-
tion of chimeric transcripts (Edgren, 2011) (Maher,
2009b) (Maher, 2009a). Chimeric transcripts cause
the production of a new protein in place of the two
original proteins that would result in absence of a
fusion. In (Maher, 2009a), short paired-end reads
have been demonstrated to allow a better identifica-
tion of fusion transcripts with respect to single long
reads, thus improving the accuracy when retrieving
the list of possible fused gene candidates. Paired-end
reads are particular reads for which only the ends of
the DNA/RNA strand are sequenced. The two ends,
also called mates of the read, are spaced by a gap
of unknown nucleotides, whose size is approximately
known. Two alternative situations might occur ac-
cording to the reads arrangement over the fusion: i)
Each mate of the read maps on a different gene of
the couple of genes involved in the fusion. The read
is then defined as
encompassing
; ii) Only a single
mate of a paired-end read overlaps the fusion junction
while the other maps on one of the two genes involved
in the fusion. The read is then considered as
spanning
the fusion boundary.
In this work we present a novel methodology for
the detection of fusion transcripts taking advantage
of both spanning and encompassing short paired-end
reads. In order to improve quality and selectivity
of fusion discovery, the framework is built on top
of an accurate gene fusion model based on validated
experimental evidence (Edgren, 2011) and leverages
upon state-of-art alignment and transcript analysis al-
gorithms (Trapnell, 2009) (Trapnell, 2010), aimed at
overcoming RNA-Seq challenges related to multiple
read alignment and novel transcript discovery.
2 CHIMERIC DETECTION FLOW
Figure 1 depicts the proposed analysis flow for the
detection of chimeric transcripts. A preliminary anal-
ysis on the paired-end samples is performed as first
step. Specifically, this phase consists of a paired end
331
Abate F., Paciello G., Acquaviva A., Ficarra E., Ferrarini A., Delledonne M. and Macii E..
A NOVEL ANALYSIS FLOW FOR FUSED TRANSCRIPTS DISCOVERY FROM PAIRED-END RNA-SEQ DATA.
DOI: 10.5220/0003789003310334
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2012), pages 331-334
ISBN: 978-989-8425-90-4
Copyright
c
2012 SCITEPRESS (Science and Technology Publications, Lda.)

Figure 1: Analysis flow for the detection of chimeric tran-
scripts.
reads mapping onto the reference genome and splic-
ing events detection. Compared to state of the art
solutions (Sboner, 2010)(McPherson, 2011), the pro-
posed methodology is built on top of TopHat (Trap-
nell, 2009), a tool for the detection of not annotated
splicing events. The alignment results are analyzed
with Cufflinks (Trapnell, 2010) in order to reveal the
transcripts expressed in the sample data set and create
an assembly model.
The adoption of TopHat (Trapnell, 2009) and Cuf-
flinks (Trapnell, 2010) allows to perform the detection
of expressed transcript without providing any annota-
tion information. As chimeric transcripts are unpre-
dictable events that might also involve new isoform
transcripts, a chimeric analysis built on top of the re-
sults of TopHat (Trapnell, 2009) and Cufflinks (Trap-
nell, 2010) is essential for an accurate detection of
fused genes. After the preliminary analysis of paired-
end samples, the proposed flow is mainly composed
of the following four steps:
Mapping Read Mates to Gene. A read encompasses
the fusion junction when the two mates maps on dif-
ferent genes. However, the results coming from the
alignment provide mainly information on the location
of the reference genome where the read maps to. In
order to determine the gene where the read matches it
is necessary to map the read location on an annotation
file. Therefore, this phase maps each aligned mate on
the transcripts detected by Cufflinks (Trapnell, 2010)
overcoming the limit of restricting the analysis only
to known and annotated transcripts.
Chimeric Candidates Detection. A set of read pairs
having the two mates mapping on two different gene
transcripts implies the presence of a gene fusion. The
Chimeric Candidates Detection phase analyzes the
set of read mates mapped on one or more transcripts
collected in the Mapping Read Mates to Gene phase.
Specifically, the subset of reads having two mates en-
compassing on different genes is selected. All those
gene couples detected by encompassing reads are re-
ported as putative candidates for a gene fusion.
Junction Breakpoint Detection. Starting from the
list of fused candidates previously detected, the scope
of this phase is to determine the exact junction break-
point for each putative chimeric candidate. Splicing
discovery programs (Trapnell, 2009)(Bryant, 2010)
overcome the classical alignment tools limitation in
the sense that they efficiently detect the exact intron-
exon boundary. The adoption of these tools instead of
canonical alignment programs results extremely use-
ful in detecting gene fusion. However, due to the con-
siderable computational complexity they are limited
in retrieving gene fusions across the entire genome
reference. Junction Breakpoint Detection overcomes
the limitation adopting a virtual reference strategy: 1)
For each couple of gene candidates a virtual refer-
ence consisting in the concatenation of the two genes
is created; 2) TopHat splicing discovery program is
launched on the virtual reference providing as input
those reads that were initially discarded in the pre-
liminary analysis. TopHat (Trapnell, 2009) aligns the
read end mates on the virtual gene fusion instead of
human genome reference. Thus, during the detection
of junction breakpoint, the read alignments must be
coherently translated from virtual to genomic coor-
dinates. Moreover, TopHat (Trapnell, 2009) analysis
reports all the mapping reads including the read mates
spanning the junction breakpoint region. Specifically,
the Junction Breakpoint Detection phase extracts for
each read the information about the location of the
start and end alignment point. If the read starting
alignment point is located before the virtual gene fu-
sion boundary and the read ending alignment point is
located after the virtual gene fusion boundary, a span-
ning read is detected. At the end of the Exact Junction
Breakpoint Analysis the set putative junctions, as well
as the supporting spanning reads, is reported for each
gene candidate.
Chimeric Candidates Validation. The previous
phases produce an extensive list of putative fused
genes. However, the detection of chimeric transcripts
can be affected by propagation errors due to both
alignment limitations and artifacts in the experimen-
tal preparation of the sample. In order to accurately
detect chimeras, the Chimeric Candidates Validation
phase selects all those fused gene candidates that
mostly fit an accurate gene fusion model detailed in
Section 3.
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
332

3 JUNCTION BOUNDARY
DETECTION
The large number of putative fused genes are filtered
according to a set of criteria reflecting an accurate
model of gene fusion. The following subsection pro-
vides the details of the most relevant criteria defining
the model.
Insert Size Coherency. In RNA-Seq paired end data,
the insert size distance is not fixed a priori and it varies
according to the specific protocol adopted in the se-
quence analysis. The distribution of the insert frag-
ment length of the aligned paired end mostly concen-
trates on a mean value with a specified standard devi-
ation. However, as emphasized in (Sboner, 2010), the
preparation of biological sample produces gene fu-
sion artifacts presenting abnormal insert size between
the sequenced ends. Therefore, in order to remove
fusion artifacts the proposed methodology estimates
the insert distance of the reads encompassing a gene
fusion candidate and removes those reads having an
insert distance size that is outlier in the fragment in-
ner size distribution.
Asymmetric Encompassing Read Distribution. As
recently investigated in (Edgren, 2011), fusions due to
PCR artifacts present an encompassing reads align-
ment that is asymmetric for the involved genes.
Specifically, it might occur that the mates encompass-
ing a fused gene are more longly aligned on one of
the two candidates whereas more concentrated in a
short range of base pairs in the corresponding gene.
In presence of asymmetric encompassing reads distri-
bution, the insert size of encompassing reads varies
around a widely variable range. Therefore, the pro-
posed methodology exploits the computation of insert
distances and it effectively removes gene fusion arti-
facts due to PCR amplification detecting asymmetric
encompassing read distribution.
Homologous Sequence Artifacts Filter. Multiple
mate matches occur due to homologies in the genome
reference. Homologous sequences affect the fusion
detection analysis because the mate pairs that nor-
mally would match on the same gene match discor-
dantly on two distinct but similar genes. Homolo-
gous region may be due both to the presence of par-
alogue genes that share long sequence regions and to
the presence of shorts similar sequences. The pro-
posed flow implements two different policies for both
cases. Concerning the long homologoussequence due
to paralogue genes a filter that query TreeFam (Li,
2006) database has been implemented. For short ho-
mologous sequences, the filter extracts and reversely
maps the read mates on the same genes. If the reads
reversely maps the gene candidates it means that the
reads encompasses the candidates due to an homolo-
gous subsequence.
Encompassing-Spanning Read Coherency. Ac-
cording to the definition of encompassing and span-
ning reads, a true gene fusion sequence results from
the consensus between encompassing and spanning
reads. If the set of encompassing and spanning reads
are located in largely different gene regions the candi-
date must be discarded an incoherent gene sequence
can be produced. Therefore, this criterion preserves
only those gene fusions with overlapping spanning
and encompassing regions.
4 RESULTS
In order to evaluate the efficiency of the proposed
flow in detecting chimeric transcripts, we analyzed
the publicly available sets of RNA-Seq data from
NCBI database (submission number SRA009053). It
is worth noting that the gene fusions occurring in
the the aforementioned data set have been validated
through RT-PCR as reported in (Berger, 2010). Ta-
ble 1 demonstrates the capability of the proposed
methodology in revealing the RT-PCR validated fu-
sions. These samples have a coverage of at most 16
million reads, a read length of 50 bp and fragment
length spanning from 350 to 500. All the 14 fusions
validated in the 7 samples of melanoma cells (Berger,
2010) have been successfully detected. Table 2 shows
some details of the detected gene fusion. In fact, for
each sample the name of the 5’ and 3’ gene are re-
ported. Moreover, the table highlights for each fu-
sion the number of encompassing and spanning reads.
This information is extremely important in the anal-
ysis of chimeric transcripts. In fact, the number of
spanning and encompassing reads across the fused
junction is directly correlated with the sequencing ex-
perimental coverage. Therefore, the proposed analy-
sis flow is able to detect the gene fusion also in case
of low coverage where the number of spanning and
encompassing reads is reduced.
Moreover, the detection of a chimeric transcript
analysis flow built on top of the TopHat and Cuf-
flinks tools represents the major novelty of the pro-
posed methodology. In fact, the adoption of TopHat
and Cufflinks allows to detect novel transcripts iso-
forms that can be recombined with known transcript
in a new chimeric gene. Therefore, in order to demon-
strate the effectiveness of the proposed flow in detect-
ing fused genes involving an unknown transcript iso-
form we report the analysis results conducted on the
sample SRR018259 (See Table 3). Specifically, the
second and third column reports the name and the ge-
A NOVEL ANALYSIS FLOW FOR FUSED TRANSCRIPTS DISCOVERY FROM PAIRED-END RNA-SEQ DATA
333
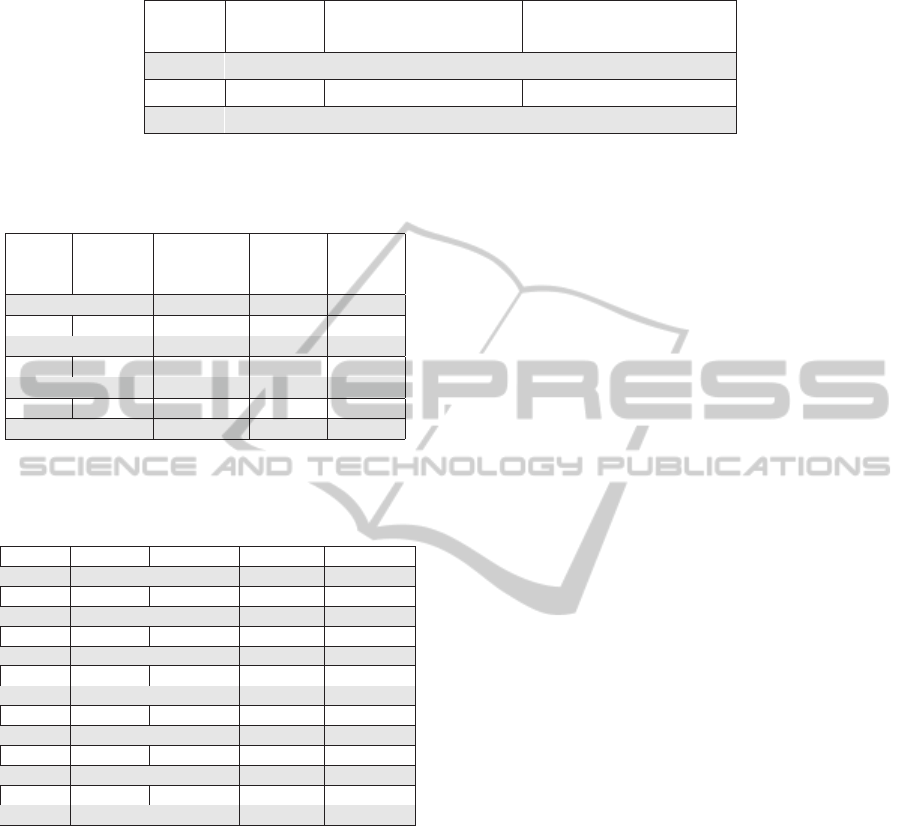
Table 3: Fusions involving unknown transcript isoform.
Library Known Genome Coordinates Genome Coordinates
Sample* Gene Known Gene Unknown Gene
018259 CCDC88C chr14:91850657-91850720 chr11:125938443-125938495
018259 PRICKLE4 chr6:41757443-41757522 chr12:125540856-125540946
018259 SLC25A1 chr11:85646172-85646214 chr22:19164633-19164667
*All the library identifiers refer to the accession number reporting the SRR prefix in the NCBI databank.
Table 1: Fusions predicted on publicly available RNA-Seq
data.
Library Reads Read Length Fragment Validated
[#] Length Predicted
(Millions) Fusions
018259 14 50 500 1
018260 16 50 500 2
018261 16 50 500 1
018265 8 50 500 1
018266 15 50 500 4
018267 15 50 500 2
018269 15 50 350 3
*All the library identifiers refer to the accession number reporting the SRR
prefix in the NCBI databank.
Table 2: Fusions detected in publicly available data set.
Library* 5’ Gene 3’ Gene Enc. Reads Span. Reads
018259 KCTD2 ARHGEF12 4 2
018260 ITM2B RB1 17 2
018260 ANKHD1 C5orf32 7 23
018261 GCN1L1 PLA2G1B 3 1
018265 WDR72 SCAMP2 2 1
018266 C1orf61 CCT3 37 25
018266 MIXL1 PARP1 5 4
018266 C11orf67 SLC12A7 40 22
018266 GNA12 SHANK2 23 6
018267 TLN1 C9orf127 14 1
018267 ALX3 RECK 21 6
018269 ABL1 BCR 89 12
018269 NUP214 XKR3 58 16
*All the library identifiers refer to the accession number reporting the SRR
prefix in the NCBI databank.
nomic coordinates of the known gene involved in the
fusion and in the fourth column we report the coor-
dinates of the unknown transcripts resulting from the
cufflinks analysis. The coordinates refers to the ge-
nomic location corresponding to the concentration of
both encompassing and spanning reads, thus referring
to the region across the fused junction breakpoint.
5 CONCLUSIONS
In this paper we presented a novel analysis flow for
the detection of chimeric transcripts in RNA-Seq data.
The proposed flow is built on top of TopHat splicing
detection tool and exploits the capability of Cufflinks
to extend the fused genes research to novel transcripts
isoforms. Moreover, the proposed methodology se-
lects those fused genes candidates that mostly fit an
accurate model of gene fusion based of experimen-
tal evidences recently reported in biomedical litera-
ture (Edgren, 2011). The experimental results demon-
strate the efficiency of the proposed flow in detect-
ing chimeric transcripts applying the methodology to
a publicly available dataset. Furthermore, we also
showed the capability of the tool in reporting fusions
involving unknown and unannotated transcript iso-
forms.
REFERENCES
Berger, M. F. (2010). Integrative analysis of the melanoma
transcriptome. Genome Research.
Bryant, D. W. J. (2010). High-throughput dna sequencing
concepts and limitations. Bioinformatics.
Edgren, H. (2011). Identification of fusion genes in breast
cancer by paired-end rna-sequencing. Genome Biol-
ogy.
Kircher, M. (2010). High-throughput dna sequencing con-
cepts and limitations. Bioessays.
Li, H. (2006). Treefam: a curated database of phyloge-
netic trees of animal gene families. Nucleic Acids Re-
search.
Magalhes, J. P. D. (2010). Next-generation sequencing in
aging research: Emerging applications, problems, pit-
falls and possible solutions. Ageing Research Review.
Maher, C. A. (2009a). Chimeric transcript discovery by
paired-end transcriptome sequencing. PNAS.
Maher, C. A. (2009b). Transcriptome sequencing to detect
gene fusions in cancer. Nature.
McPherson, A. (2011). defuse: An algorithm for gene fu-
sion discovery in tumor rna-seq data. PLoS Computa-
tional Biology.
Sboner, A. (2010). Fusionseq: a modular framework for
finding gene fusions by analyzing paired-end rna-
sequencing data. Genome Biology.
Trapnell, C. (2009). Tophat: discovering splice junctions
with rna-seq. Bioinformatics.
Trapnell, C. (2010). Transcript assembly and quantification
by rna-seq reveals unannotated transcripts and isoform
switching during cell differentiation. Nature Biotech-
nology.
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
334
