
SEMI-SUPERVISED LEARNING OF ALTERNATIVELY SPLICED
EXONS USING EXPECTATION MAXIMIZATION TYPE
APPROACHES
Ana Stanescu and Doina Caragea
Computing and Information Sciences, Kansas State University, Manhattan, KS, U.S.A.
Keywords:
Semi-supervised learning, Expectation maximization, Alternative splicing.
Abstract:
Successful advances in DNA sequencing technologies have made it possible to obtain tremendous amounts
of data fast and inexpensively. As a consequence, the afferent genome annotation has become the bottleneck
in our understanding of genes and their functions. Traditionally, data from biological domains have been an-
alyzed using supervised learning techniques. However, given the large amounts of unlabeled genomics data
available, together with small amounts of labeled data, the use of semi-supervised learning algorithms is de-
sirable. Our purpose is to study the applicability of semi-supervised learning frameworks to DNA prediction
problems, with focus on alternative splicing, a natural biological process that contributes to protein diversity.
More specifically, we address the problem of predicting alternatively spliced exons. To utilize the unlabeled
data, we train classifiers via the Expectation Maximization method and variants of this method. The experi-
ments conducted show an increase in the quality of the prediction models when unlabeled data is used in the
training phase, as compared to supervised prediction models which do not make use of the unlabeled data.
1 INTRODUCTION
Over the last decade, major advancements in the next
generation sequencing technologies have led to an un-
precedented growth in the volume of biological data,
which is now acquired with high speed and low costs.
As the emphasis progressively switches from data
generation to data interpretation (Baldi and Brunak,
2001), the annotation process relies more and more on
automated systems. Many genome annotation tasks
can be formalized as supervised classification prob-
lems where a learning classifier system is trained to
produce the best prediction: it learns from observed
instances (a.k.a., labeled data) to make predictions re-
garding new unseen instances (a.k.a., unlabeled data).
For example, labeled instances such as recognized
splice sites, or laboratory established protein func-
tions, can be used to train the classifier, which is sub-
sequently used to categorize new instances for which
such information is still unknown.
Supervised machine learning techniques have
been successfully used for many problems in the field
of bioinformatics (Zhang and Rajapakse, 2009) but
their effectiveness relies on the availability of labeled
data in large amounts. Obtaining labeled data remains
a barrier, as it is a slow and expensive process, which
usually requires human effort, while large amounts
of unlabeled instances are easily available. A branch
of machine learning, called semi-supervised learning
(SSL), advocates the use of unlabeled data to improve
classifiers learned from small amounts of labeled data
only when large amounts of unlabeled data are avail-
able. SSL approaches have shown great potential in
various domains, such as text (Nigam et al., 2000;
Dai et al., 2007) and image classification (Rosenberg
et al., 2005), sentiment categorization (Goldberg and
Zhu, 2006), natural language processing (Collins and
Singer, 1999), yet have not been applied to a great
extent in bioinformatics, where most prominent ex-
ceptions are related to protein analyses (Weston et al.,
2006; Kall et al., 2007). The aim of this study is to
evaluate the suitability of SSL techniques for DNA
sequence classification, with focus on predicting al-
ternative splicing events.
Alternative (or differential) splicing was first ob-
served in the late 1970’s (Chow et al., 1977) and
was speculated to be an exceptional occurrence.
Since then, due to its omnipresence in all eukaryotic
genomes (Black, 2003), it has been acknowledged as
a natural phenomenon: if its pre-mRNA is alterna-
tively spliced, a gene can encode more than one pro-
tein. Alternative splicing usually takes place after
240
Stanescu A. and Caragea D..
SEMI-SUPERVISED LEARNING OF ALTERNATIVELY SPLICED EXONS USING EXPECTATION MAXIMIZATION TYPE APPROACHES.
DOI: 10.5220/0003791802400245
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2012), pages 240-245
ISBN: 978-989-8425-90-4
Copyright
c
2012 SCITEPRESS (Science and Technology Publications, Lda.)

transcription (of the pre-messenger RNA from DNA)
and right before mRNA translation, giving rise to sev-
eral transcripts (or splice variants), which in turn en-
code different polypeptides, making a gene highly ef-
ficient with respect to the proteome formation.
There are a few manifestations of this phe-
nomenon, some in which exons are spliced out and
others where introns are retained. Our study is fo-
cused on the prediction of alternatively spliced ex-
ons. Exons that are not alternatively spliced are called
constitutive. Thus, we will address the task of dis-
criminating between alternatively spliced exons and
constitutive exons by representing this task as a bi-
nary (yes/no) classification problem. We learn prob-
abilistic label Na
¨
ıve Bayes (Nigam et al., 2000) and
Support Vector Machine (SVM) (Vapnik, 1995) clas-
sifiers from a combination of labeled and unlabeled
data sets using expectation maximization type ap-
proaches in a semi-supervised framework. The main
contribution of our work is experimental and it shows
that semi-supervised approaches, which employ the
expectation maximization technique, are effective at
exploiting the unlabeled biological data.
2 RELATED WORK
The Expectation Maximization technique (EM) origi-
nates from statistics and was later formalized (Demp-
ster et al., 1977) as an iterative algorithm for maxi-
mum likelihood estimation. Its applicability to learn-
ing probability distributions and capability of utiliz-
ing sufficiently large amounts of unlabeled data in or-
der to build and improve upon a model makes it a very
powerful technique which has gained a lot of popu-
larity in the field of machine learning. It has been
shown to perform well in text classification problems
(Nigam et al., 2000). In biological and medical do-
mains, the EM has been used for modeling data for
creating protein profiles (Nesvizhskii et al., 2003),
for finding motifs within sequences (Lawrence and
Reilly, 1990), for image reconstruction through clus-
tering (Lawrence and Reilly, 1990), etc. More re-
cently, in machine learning applications, it has been
found very useful in semi-supervised frameworks, for
text classification (Nigam et al., 2000), audio catego-
rization tasks (Moreno and Agarwal, 2003), and im-
age retrieval (Dong and Bhanu, 2003).
Among others, a semi-supervised approach us-
ing EM and Na
¨
ıve Bayes with Probabilistic Labels
was proposed by Nigam et al. (2000) in the context
of text classification. Their results on three differ-
ent text corpora show dramatic improvements when
large amounts of unlabeled data are used together
with small amounts of labeled data. We will study
this algorithm and some of its variants in the context
of predicting alternatively spliced exons.
Given our application problem, work on identify-
ing alternatively spliced exons in genomic sequences
is also relevant to the work presented. Tradition-
ally, this type of problem has been solved by con-
ducting wet-lab experiments. As lab work is very
tedious, computational methods which use the align-
ment of Expressed Sequence Tags (EST) to genome
have emerged (Nagaraj et al., 2007). More recently,
prediction of alternative splicing has been the focus of
machine learning research work which makes use of
Support Vector Machines (Dror et al., 2005; Ratsch
et al., 2005) to produce fast and accurate classifiers.
Specialized kernels that model similarities between
sequences are used in these studies.
To the best of our knowledge, SSL techniques
using the EM algorithm have not been applied to
the problem of predicting alternatively spliced exons.
The work presented in this paper shows that these
types of approaches constitute a promising direction.
3 DATA AND FEATURES
The dataset used in our experiments is made avail-
able online by the Friedrich Miescher Laboratory of
the Max Planck Society (T
¨
ubingen, Germany), at
the URL: http://www.fml.tuebingen.mpg.de/raetsch/
suppl/RASE/data sets. It contains 3018 DNA se-
quences from the nematode C. elegans. Each com-
prising one exon along with its left and right flank-
ing introns. In short, R
¨
atsch et al. generated these
instances by aligning expressed sequence tags (EST)
against genomic DNA. This modus operandi pro-
duced 2531 constitutive exons and 487 alternatively
spliced exons. The data set has been previously used
by the aforementioned authors in the context of super-
vised learning (Ratsch et al., 2005).
It is known that regulatory elements located both
in introns or exons can influence alternative splic-
ing (Chasin, 2007). Such regulatory sequences can
be identified as motifs. In biology, a motif is usu-
ally defined as a short and widespread nucleotide
(or amino-acids) sequence pattern that captures some
commonalities between related sequences, thus hav-
ing a prevalent biological significance. We consider
both intronic motifs (a.k.a., intronic regulatory se-
quences) and exonic motifs (a.k.a., exonic splicing
enhancers) to represent our instances as feature vec-
tors. More precisely, we convert each sequence into
a vector, where each dimension corresponds to a mo-
tif, and each value is given by the motif’s frequency
SEMI-SUPERVISED LEARNING OF ALTERNATIVELY SPLICED EXONS USING EXPECTATION
MAXIMIZATION TYPE APPROACHES
241

(count). It is also known that the lengths of an exon
and its flanking introns are discriminative (Dror et al.,
2005) with respect to the problem of predicting if the
exon is alternatively spliced or constitutive. Thus, an
additional set of features used in our work are ob-
tained from lengths.
For our first set of features, we use the Intronic
Regulatory Sequences (IRS) established by compara-
tive genomics in Nematodes by Kabat et al. Briefly,
the introns that flank alternatively spliced exons show
evidence of high nucleotide preservation, leading to
the identification of similar k-mers between C. ele-
gans and C. briggsae. Kabat et al. (2006) provide
the description of conserved and non-conserved pen-
tamers and hexamers from the upstream and down-
stream introns. Among these, 165 motifs are iden-
tified in our sequences (using simply scanning) and
therefore used as a feature set subsequently.
The second feature set was obtained using the
method from (Pertea et al., 2007). It consists of 45
Exonic Splicing Enhancers (ESEs). ESEs direct or
enhance accurate splicing of pre-mRNA into messen-
ger RNA; they are usually 6 nucleotides long.
We used the length features (LF) from (Ratsch
et al., 2005). Specifically, the length of each up-
stream intron, exon and downstream intron (of ev-
ery sequence in the set), was used to generate 30-
dimensional logarithmically spaced vectors, for a to-
tal of 90 features per instance (corresponding to the 3
lengths). Within the same group, we also included a
set of 3D vectors characterizing the frame of the stop
codon (which together results in 15 more features).
Ultimately, we have 315 features based on motifs,
length and frame of the stop codon. The labels of the
instances were not used when generating features.
4 APPROACHES
EM is a probabilistic algorithm which allows the
learning of a model in the presence of missing data,
through iterative parameter estimation. The EM algo-
rithm consists of two steps: (1) The Expectation step,
to fill in the missing data: in our context, the class la-
bels of the unlabeled data, and (2) the Maximization
step, to calculate a maximum a posteriori estimate for
the model parameters.
In a semi-supervised setup, EM can be put into
practice as follows: a classifier is initially trained
with just the labeled data (1). It is then used to clas-
sify the unlabeled data (2). Next, all the data (i.e.,
originally labeled data along with newly classified in-
stances from the unlabeled set) is used to train a new
classifier (3). Steps 2 and 3 iterate until convergence.
Although EM might look like a heuristic method,
it does have a rigorous foundation. It is guaranteed to
find a local optimum of data likelihood (Wu, 1983).
In this paper, for the problem of predicting alternative
splicing in a semi-supervised mode, we first use the
EM technique with a generative model as base classi-
fier, namely Na
¨
ıve Bayes (Nigam et al., 2000). Sec-
ond, we also explore EM with a discriminative ap-
proach, Support Vector Machines (SVM), as the base
classifier (Brefeld and Scheffer, 2004).
4.1 SSL using EM and NBM
As described above, the usage of EM in a semi-
supervised framework assumes that a classifier is first
learned from the originally labeled data. Given that
our data has partly a motif count representation, we
learn a Na
¨
ıve Bayes Multinomial (NBM) classifier
from the motif representation of the labeled data.
Note that we use the multinomial model to capture
the frequency of a motif, rather than just its pres-
ence or absence, which would require a multi-variate
Bernoulli event model (McCallum and Nigam, 1998).
Following the notation from (Nigam et al., 2000), we
use θ to denote the model parameters and D to rep-
resent the data. Learning the model is equivalent to
finding θ that maximizes the log of the posterior prob-
ability P(θ|D). This is equivalent to finding θ that
maximizes log[P(θ) · P(D|θ)]. Next, we use the re-
sulting model to soft-label the instances in the unla-
beled set by assigning them probabilistic class labels.
For each instance in the unlabeled data set we get a
probability distribution over the two classes and use
this distribution to compute fractional counts, mean-
ing that the actual counts in a class are proportional to
the corresponding class probability of that example.
With this new model, we re-label the unlabeled
sequences. This process can be repeated for a fixed
number of steps or until convergence, i.e., the la-
bels from one iteration are very similar to the ones
in the previous iteration. One variation of the EM ap-
proach can be obtained by assigning different weights
to the labeled and unlabeled instances when learning
the NBM (Nigam et al., 2000). This can be achieved
by introducing a new weighting factor which controls
the weight of each newly classified unlabeled exam-
ple, thus adjusting (decreasing) the influence of the
unlabeled data over the model and granting more au-
thority to the labeled examples. For this model we use
the formula from (Nigam et al., 2000) where z
i j
is 0
or 1 for the labeled instances (depending on their ac-
tual class) or P(c
j
|d
i
) for the unlabeled instances and
C is the set of classes – in our case, positive (1) or
negative (0) – and d
i
an instance in the labeled data
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
242

set D; when w = 1, the algorithm is identical to the
one described previously:
log(P(θ)) +
∑
d
i
∈D
|C|
∑
j=1
z
i j
log(P(c
j
|θ)P(d
i
|c
j
;θ))
+w(
∑
d
i
∈D
|C|
∑
j=1
z
i j
log(P(c
j
|θ)P(d
i
|c
j
;θ))) (1)
Another popular SSL algorithm is self-training
(aka self-teaching or bootstrapping). It was intro-
duced in (Yarowsky, 1995) where it was used success-
fully in a natural language processing problem. Char-
acterized as a hybrid between EM and Co-Training
(Nigam and Ghani, 2000), it can be used with any
base-classifier to pull more training cases from the
unlabeled set. However, unlike EM which uses all
predictions to update the parameters of its model,
self-training only uses the best predictions at each
round and disregards the instances which are labeled
with low confidence. Unlike Co-Training (Blum and
Mitchell, 1998), it is a single-view learning algorithm.
An important condition is to maintain the ratio of pos-
itive to negative examples across datasets.
4.2 SSL using EM with SVM
Support Vector Machines (SVM) represent a rela-
tively recent family of supervised learning methods
that can be applied to binary classification problems,
generally yielding very accurate results. Given their
popularity, we also use SVM as a base classifier in
the above described EM procedure, with a Gaussian
kernel and an error cost C = 0.5. Just like in the case
of NBM, we use the weighting scheme for SVM as
well. Each newly classified instance from the un-
labeled data set is further used in retraining with a
weight coefficient w. We denote each experiment by
NBMemW(w) and SVMemW(w), where the weight
w ∈ {0.01, 0.1, 0.25, 0.3, 0.5, 0.75}. The self-training
implementation is similar to the one using NBM de-
scribed in Section 4.1. They are indicated as NBM-
self(s,i) and SVMself(s,i) where s is the sample size
and i is the number of iterations.
5 EXPERIMENTAL SETUP
An objective evaluation of any predictive model re-
quires the use of the cross validation technique. To
estimate how well our classifiers will generalize to
new data, and to maintain the trend set by (Ratsch
et al., 2005), we employed 5-fold cross validation.
We then split the training set into labeled and unla-
beled subsets of different sizes. The unlabeled sub-
set was simply obtained by intentionally ignoring the
label information. Given that our data is skewed
– we have approximately five times more instances
labeled as ”constitutive” than we do ”alternatively
spliced”, and so measuring the accuracy of the pre-
dictions would not reflect the true value of our clas-
sifier (Provost et al., 1998), we have reported the
performance in terms of area under the ROC curve
(AUC)(Huang and Ling, 2005).
In order to assess the behavior of our SSL al-
gorithms, we compare their performance against the
lower and upper bounds of each experiment, in terms
of AUC values. These values will give us an indica-
tion of how much improvement, if any, there can be
expected from using the unlabeled data in a particu-
lar case (i.e., for a particular algorithm and a set of
motifs). First, we run a supervised version of the al-
gorithms, maintaining the same folds, but assuming
no data in the training set to be unlabeled. Recall that
we deliberately treat some instances as unlabeled to
simulate the semi-supervised environment and to be
able to judge out results. This value mainly tells how
good the set of motifs really is and gives an upper
limit for how well we can anticipate to do in the semi-
supervised framework. Learning just from the labeled
subset will give us a lower bound of performance.
6 RESULTS
The first experiment involves the NBM classifier with
fractional labels, along with IRS and ESE motifs. The
use of LF is not justified in this setup, as the values
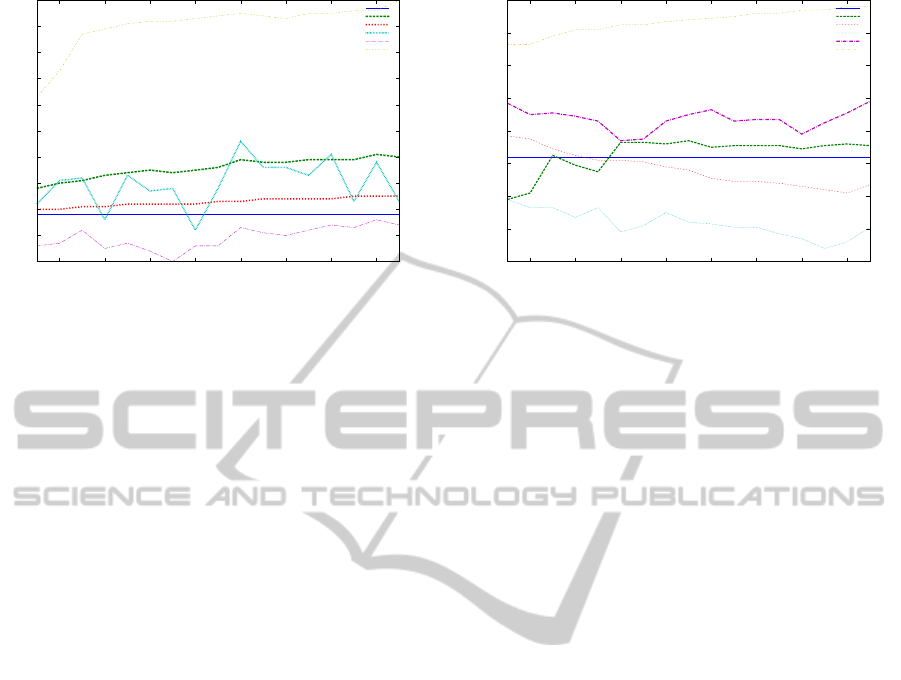
are not fit for a multinomial model. Figure 1 shows
the performance of the classifier when trained on 5%
of the labeled data along with different amounts of
unlabeled data, varying from 15% to 95%. For the
lower bound (LB), the classifier was trained only on
5% of the labeled data (approximately 120 examples).
It has been observed that when given a weight greater
than 0.5, the unlabeled data adds noise, resulting in
a performance poorer than the LB. The same trend is
maintained when the amount of labeled data is varied
from 5% to 30% while the unlabeled data is fixed at
70%: NBMem(0.1) gives the best results, followed by
NBMem(0.25), NBMem(0.3) and degrading towards
NBMem(1.0). In practice, is not always the case for
the unlabeled data to match the assumptions made by
the generative model, leading to a degradation of the
EM performance (Nigam et al., 2000); this could be
one possible explanation for our DNA data set, since
the EM with NBM implementation outperforms the
SEMI-SUPERVISED LEARNING OF ALTERNATIVELY SPLICED EXONS USING EXPECTATION
MAXIMIZATION TYPE APPROACHES
243
0.83
0.84
0.85
0.86
0.87
0.88
0.89
0.9
0.91
0.92
0.93
0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
AUC value
Amount of Unlabeled Data
Lower Bound
NBMem (0.1)
NBMem(0.01)
NBMself(200,200)
NBMem (1.0)
Upper Bound
Figure 1: EM and self-training NBM performance with IRS
and ESE motifs when varying the amount of labeled data.
LB only when the contribution of the unlabeled data
is diminished. This suggests that enforcement upon
the influence of the unlabeled data during training is
useful but if too little importance is given (w = 0.01)
some valuable information remains unexploited. Fur-
thermore, the learned model betters with the increase
in the amount of unlabeled data; so probably if more
unlabeled data is added, the quality will continue to
grow. This hypothesis is worth investigating further
in future work. For the self-training approach we have
set a growth size (i.e., number of instances to be added
to the labeled set at each iteration) of 6, such that the
class ratio (5:1) is maintained. We varied the sample
size (i.e., how many examples are classified per iter-
ation amongst which the best 6 will be added to the
labeled set) between 50 and 200 and the number of
iterations from 50 to 200. The best scores on average
were achieved for 200 sample size and 200 iterations.
With the SVM implementation of the EM algo-
rithm, the LF can be included. Figure 2 represents
experiments for EM and SVM using IRS, ESE motifs
and also LF. Although there is not much improvement
over the baseline, a weight of 0.1 is still better than all
the other weighting values, however, self-training out-
performs all weights as well as the baseline.
Variations in terms of AUC when the model is
learned from increasing amounts of labeled data while
keeping the amount of unlabeled data fixed to 70%,
show that for the SVM classifier, self-training per-
forms better than the EM variation with weights in
this context too, however the results do not go beyond
the LB. In a strictly supervised setup, NBM achieves
the highest AUC value overall (0.93), followed by
SVM using IRS and ESE motifs (0.921) and SVM
using IRS, ESE and the LF (0.916).
0.76
0.78
0.8
0.82
0.84
0.86
0.88
0.9
0.92
0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
AUC value
Amount of Unlabeled Data
Lower Bound
SVMemW (0.1)
SVMemW(0.01)
SVMemW (1.0)
SVMself(50,25)
Upper Bound
Figure 2: EM and self-training SVM performance with IRS,
ESE and LF when varying the amount of labeled data.
7 CONCLUSIONS
This work represents an empirical study of EM type
algorithms in the context of SSL applied to the clas-
sification of DNA sequences, using NBM and SVM
as base classifiers. We have shown that unlabeled
data does help improve the quality of the predictions
when the influence it has over the model in the train-
ing phase is small. In the case of NBM with proba-
bilistic labels, the IRS and ESE motifs are sufficient to
boost the performance over the LBs; when unlabeled
data is added, the predictions improve gradually. For
SVM as base classifier in the EM framework, in addi-
tion to the weighting scheme, self-training also shows
promising results. As expected, over all experiments,
predictions improve with the increase of labeled data
in the training phase. We can also conclude that NBM
is most effective in the supervised framework when
using IRS and ESE motifs.
8 FUTURE WORK
Many aspects that are critical to alternative splicing
classification in a semi-supervised setup still need to
be explored: from using more unlabeled data and
more powerful discriminative motifs to feature selec-
tion, parameter fine-tuning via validation setups and
exploring new semi-supervised approaches. Given
that large margin classification yields state-of-the-art
results for many prediction problems, including alter-
native splicing (Ratsch et al., 2005), it is definitely
worth investigating the idea of support vector ma-
chines with specialized kernels, (i.e., kernels for com-
putational biology (Ben-Hur et al., 2008)) in a trans-
ductive (Gammerman et al., 1998) manner as well.
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
244

REFERENCES
Baldi, P. and Brunak, S. (2001). Bioinformatics: the ma-
chine learning approach. MIT Press.
Ben-Hur, A., Ong, C. S., Sonnenburg, S., Scholkopf, B.,
and Ratsch, G. (2008). Support vector machines and
kernels for computational biology. PLoS computa-
tional biology.
Black, D. L. (2003). Mechanisms of alternative pre-
messenger RNA splicing. Annual Review of Biochem-
istry.
Blum, A. and Mitchell, T. (1998). Combining labeled
and unlabeled data with Co-Training. In Proceedings
of the eleventh annual conference on Computational
learning theory. ACM.
Brefeld, U. and Scheffer, T. (2004). Co-EM support vec-
tor learning. In In Proceedings of the International
Conference on Machine Learning.
Chasin, L. A. (2007). Searching for splicing motifs. Ad-
vances in Experimental Medicine and Biology.
Chow, L. T., Gelinas, R. E., Broker, T. R., and Roberts, R. J.
(1977). An amazing sequence arrangement at the 5’
ends of adenovirus 2 messenger RNA. Cell.
Collins, M. and Singer, Y. (1999). Unsupervised models
for named entity classification. In In Proceedings of
the Joint SIGDAT Conference on Empirical Methods
in Natural Language Processing and Very Large Cor-
pora.
Dai, W., Xue, G., Yang, Q., and Yu, Y. (2007). Transfer-
ring naive bayes classifiers for text classification. In
In Proceedings of the 22nd AAAI Conference on Arti-
ficial Intelligence.
Dempster, A. P., Laird, N. M., and Rubin, D. B. (1977).
Maximum likelihood from incomplete data via the
EM algorithm. Journal of the Royal Statistical So-
ciety.
Dong, A. and Bhanu, B. (2003). A new semi-supervised
EM algorithm for image retrieval. Computer Vision
and Pattern Recognition.
Dror, G., Sorek, R., and Shamir, R. (2005). Accurate iden-
tification of alternatively spliced exons using support
vector machine. Bioinformatics (Oxford, England).
Gammerman, A., Vovk, V., and Vapnik, V. (1998). Learning
by transduction. In In Uncertainty in Artificial Intelli-
gence. Morgan Kaufmann.
Goldberg, A. B. and Zhu, X. (2006). Seeing stars when
there aren’t many stars: graph-based semi-supervised
learning for sentiment categorization. In Proceedings
of the First Workshop on Graph Based Methods for
Natural Language Processing. Association for Com-
putational Linguistics.
Huang, J. and Ling, C. X. (2005). Using a u c and accuracy
in evaluating learning algorithms. IEEE Transactions
on Knowledge and Data Engineering.
Kabat, J. L., Barberan-Soler, S., McKenna, P., Clawson, H.,
Farrer, T., and Zahler, A. M. (2006). Intronic alterna-
tive splicing regulators identified by comparative ge-
nomics in nematodes. PLoS computational biology.
Kall, L., Canterbury, J. D., Weston, J., Noble, W. S., and
MacCoss, M. J. (2007). Semi-supervised learning
for peptide identification from shotgun proteomics
datasets. Nature methods.
Lawrence, C. E. and Reilly, A. A. (1990). An expec-
tation maximization (EM) algorithm for the identifi-
cation and characterization of common sites in un-
aligned biopolymer sequences. Proteins.
McCallum, A. and Nigam, K. (1998). A comparison of
event models for naive bayes text classification. Di-
mension Contemporary German Arts And Letters.
Moreno, P. J. and Agarwal, S. (2003). An experimental
study of semi-supervised EM. Technical report, HP
Labs.
Nagaraj, S. H., Gasser, R. B., and Ranganathan, S. (2007).
A hitchhiker’s guide to expressed sequence tag (est)
analysis. Briefings in bioinformatics.
Nesvizhskii, A. I., Keller, A., Kolker, E., and Aebersold, R.
(2003). A statistical model for identifying proteins by
tandem mass spectrometry. Analytical Chemistry.
Nigam, K. and Ghani, R. (2000). Analyzing the effective-
ness and applicability of Co-Training. In Proceedings
of the 9th International Conference on Information
and Knowledge Management. ACM.
Nigam, K., McCallum, A. K., Thrun, S., and Mitchell, T.
(2000). Text classification from labeled and unlabeled
documents using EM. Machine Learning.
Pertea, M., Mount, S. M., and Salzberg, S. L. (2007). A
computational survey of candidate exonic splicing en-
hancer motifs in the model plant Arabidopsis thaliana.
BMC bioinformatics.
Provost, F. J., Fawcett, T., and Kohavi, R. (1998). The
case against accuracy estimation for comparing induc-
tion algorithms. In Proceedings of the Fifteenth Inter-
national Conference on Machine Learning. Morgan
Kaufmann Publishers Inc.
Ratsch, G., Sonnenburg, S., and Scholkopf, B. (2005).
Rase: recognition of alternatively spliced exons in
C.elegans. Bioinformatics (Oxford, England).
Rosenberg, C., Hebert, M., and Schneiderman, H. (2005).
Semi-supervised self-training of object detection
models. In Proceedings of the Seventh IEEE Work-
shops on Application of Computer Vision. IEEE Com-
puter Society.
Vapnik, V. N. (1995). The nature of statistical learning the-
ory. Springer-Verlag New York, Inc.
Weston, J., Kuang, R., Leslie, C., and Noble, W. (2006).
Protein ranking by semi-supervised network propaga-
tion. BMC Bioinformatics.
Wu, C. F. J. (1983). On the convergence properties of the
EM algorithm. Annals of Statistics, Vol. 11, No. 1.
Yarowsky, D. (1995). Unsupervised word sense disam-
biguation rivaling supervised methods. In Proceed-
ings of the 33rd annual meeting on Association for
Computational Linguistics.
Zhang, Y.-Q. and Rajapakse, J. C. (2009). Machine learning
in bioinformatics. Wiley.
SEMI-SUPERVISED LEARNING OF ALTERNATIVELY SPLICED EXONS USING EXPECTATION
MAXIMIZATION TYPE APPROACHES
245
