
SCAFFOLD HUNTER
Visual Analysis of Chemical Compound Databases
Karsten Klein, Nils Kriege and Petra Mutzel
Department of Computer Science, Technische Universit
¨
at Dortmund, Dortmund, Germany
Keywords:
Scaffold Tree, Chemical Space, Chemical Compound Data, Integrative Visualization, Interactive Exploration.
Abstract:
We describe Scaffold Hunter, an interactive software tool for the exploration and analysis of chemical com-
pound databases. Scaffold Hunter allows to explore the chemical space spanned by a compound database,
fosters intuitive recognition of complex structural and bioactivity relationships, and helps to identify inter-
esting compound classes with a desired bioactivity. Thus, the tool supports chemists during the complex
and time-consuming drug discovery process to gain additional knowledge and to focus on regions of interest,
facilitating the search for promising drug candidates.
1 INTRODUCTION
The search for a potential new drug is often com-
pared to “searching a needle in a haystack”. This
refers to the fact that within the huge chemical space
of synthesizable small organic compounds (approxi-
mately 10
60
molecules), there is only a small fraction
of potentially active compounds of interest for fur-
ther investigation. Due to the cost and effort involved
in synthesis and experimental evaluation of potential
drugs, efficient identification of promising test com-
pounds is of utmost importance. However, orienta-
tion within chemical space is difficult, as on the one
hand there is only partial knowledge about molecule
properties, and on the other hand a large number of
potentially relevant annotations exist, as physical and
chemical properties, target information, side effects,
patent status, and many more. Some of these an-
notations also may be either predicted with a cer-
tain confidence or result from experiments, with un-
certainty and sometimes even contradicting informa-
tion. Nonetheless, there are some approaches to clas-
sify and cluster compounds for navigation. A number
of properties might be good indicators for drug-like
molecule characteristics, as, e.g., biological activity,
and there are several physico-chemical properties that
allow to discard molecules, as, e.g., stability and syn-
thesizability.
The classical drug discovery pipeline, which aims
at detecting small molecules that bind to biological
target molecules involved in a disease process (e.g.,
proteins), does not only require a large amount of ti-
me, money, and other resources, but also suffers from
a small and even decreasing success rate. Since the
behavior and impact of a chemical compound often
cannot be easily predicted or derived from simple
molecular properties, the drug discovery pipeline in-
volves high throughput screenings of large substance
libraries with millions of compounds in the early
stages to identify potentially active molecules. The
results of a screening only give an incomplete pic-
ture on a restricted area of the possible solution space,
and hence need to be analyzed to detect potential lead
structures that can be used as the starting point of the
further drug development.
As a result, the drug discovery process involves
decisions based on expertise and intuition of the expe-
rienced chemist that cannot be replaced by automatic
processes. Nonetheless this process can be greatly
supported by computational analysis methods, an in-
tuitive representation of the available data, and by
navigation approaches that allow for organized ex-
ploration of chemical space. The chemist’s workflow
therefore can be supported by automatic identification
of regions within the chemical space that may contain
good candidates with high probability and by enrich-
ing the navigation with pointers to these region within
a visual exploration and analysis process.
Even though the use of automated high throughput
methods for screening and synthesis led to large com-
pound libraries and a huge amount of corresponding
data in pharmaceutical companies and academic insti-
tutions, this did not lead to a significant increase in the
success rate. Sharing data among these actors might
626
Klein K., Kriege N. and Mutzel P..
SCAFFOLD HUNTER - Visual Analysis of Chemical Compound Databases.
DOI: 10.5220/0003836806260635
In Proceedings of the International Conference on Computer Graphics Theory and Applications (IVAPP-2012), pages 626-635
ISBN: 978-989-8565-02-0
Copyright
c
2012 SCITEPRESS (Science and Technology Publications, Lda.)

help to improve the understanding and therefore also
the discovery process. Consequently, more and more
data is made publicly available over a large number of
online databases, and computational methods to ana-
lyze the data are used to an increasing extent. How-
ever, without adequate methods to integrate and ex-
plore the data, this wealth of possibly relevant infor-
mation may even complicate the drug discovery pro-
cess. In addition, information is spread across many
resources, having different access interfaces, and even
the unambiguous identification of compounds can be
non-trivial. Integration of these data resources in a vi-
sual analysis tool with an intuitive navigation concept
facilitates drug discovery processes to a large extent.
Scaffold Hunter is a software tool for the explo-
ration and analysis of chemical compound databases
that supports the chemist in the search for drug can-
didates out of the structural space spanned by a pos-
sibly large pool of compounds. It allows navigation
in this chemical space with the help of a hierarchi-
cal classification based on compound structure, and
integrates a variety of views with appropriate analy-
sis methods. The views provide innovative graphical
visualizations as well as established representations
for data and analysis results. Combined with suit-
able interaction techniques, these components allow
to assess the chemical data with respect to the various
aspects of multidimensional data annotations in an in-
tegrated fashion. In addition, Scaffold Hunter allows
to integrate data from multiple resources and formats
over a flexible import plugin interface.
Scaffold Hunter was implemented as a prototype
application in 2007, being the first tool that allows
to navigate in the hierarchical chemical space de-
fined by the scaffold tree (Schuffenhauer et al., 2007).
The Scaffold Hunter prototype was successfully used
in an experimental study that focused on the chem-
ical aspects of using brachiation along scaffold tree
branches, proving the effectiveness of the approach
and the usefulness of our implementation (Wetzel
et al., 2009). Here, we focus on the visualization and
analysis techniques used, including new views and a
data integration concept, and on their interplay.
1.1 Related Work
Compared to other application areas, especially biol-
ogy, the support of the analysis workflow in chem-
istry by integrated tools that combine both advanced
interactive visualization as well as analysis methods
is rather weak even though the need for such tools has
been formulated quite often (IMI, 2009; Irwin, 2009).
On the one hand tools based on a data pipelining con-
cept like KNIME (Berthold et al., 2007), which feat-
ures several cheminformatics extensions, or the com-
mercial product Accelrys Pipeline Pilot are applied.
Although these approaches are more intuitive to use
than cheminformatics software libraries, they nev-
ertheless require a fair amount of expert knowl-
edge in cheminformatics and lack integrated visual
analysis concepts. On the other hand general pur-
pose visualization tools like TIBCO Spotfire are
used. Spotfire can be extended by a structure depic-
tion plugin, but lacks sophisticated domain specific
analysis methods. Concepts to classify molecules,
e.g. based on clustering by common substructures,
have first been proposed several years ago (Schuf-
fenhauer and Varin, 2011), but are often not sup-
ported in interactive visualization software. Re-
cently there have been attempts to create software
to remedy the situation: The server-based tool Mol-
wind (Herhaus et al., 2009) has been developed by re-
searchers at Merck-Serono and was inspired by Scaf-
fold Hunter. While also based on the scaffold tree
concept, Molwind uses NASA’s World Wind engine
to map scaffolds to geospatial layers. The applica-
tion SARANEA (Lounkine et al., 2010) focuses on
the visualization of structure-activity and structure-
selectivity relationships by means of “network-like
similarity graphs”, but misses a structural classifica-
tion scheme which is advisable for large data sets.
Compared to these approaches, the web based tool
iPHACE (Garcia-Serna et al., 2010) introduces basic
additional features for visual analysis, namely inter-
action heat maps, to focus on the drug-target interac-
tions. Another recent approach to support the analysis
of chemical data sets is Scaffold Explorer (Agrafiotis
and Wiener, 2010), which allows the user to define the
scaffolds with respect to his task-specific needs, but is
targeted more towards the analysis of small data sets.
Although these first approaches received a pos-
itive feedback from the pharmaceutical community,
they are more or less in a prototypical stage with
a small user base. The most likely explanation is
that chemists first need to familiarize with such ap-
proaches, as there have not been established ways for
the integrated visual analysis of chemical data so far.
1.2 Goals and Challenges
Our main goal in the development of Scaffold Hunter
was to facilitate the interactive exploration of chem-
ical space in an intuitive way also suitable for non-
experts in cheminformatics. We wanted to develop a
software tool that integrates drug discovery data and
allows to browse through the structures and data in an
interactive visual analysis approach.
Several goals guided the design and implementa-
SCAFFOLD HUNTER - Visual Analysis of Chemical Compound Databases
627

tion of Scaffold Hunter:
• The user should be able to integrate data from
public resources and from his own compound
databases.
• Views that represent a space of chemical com-
pounds in an intuitive fashion for chemists should
be automatically created.
• Interaction with the views should be possible to
adapt them to the needs of a specific task, and to
allow an analysis of the underlying data.
• Guided navigation within the compound space
should be possible, to focus on regions of interest
and to drill down to promising drug candidates.
When these goals are satisfied, the tool enables
a visual analysis workflow that supports the efficient
identification of drug candidates based on the com-
bined information available. See Figure 1 for a model
of this workflow.
Several challenges make a straightforward realiza-
tion of these goals difficult:
• The set of chemical compounds under investiga-
tion may contain several million compounds, rais-
ing both efficiency and visualization problems.
• There is a large number of potentially interesting
data annotations per compound, but the knowl-
edge on them is incomplete, and the relation be-
tween molecular properties and the biological ef-
fects are complex and difficult to characterize.
• In order to take advantage of publicly available
information, including large online databases as
PubChem, Zinc, or ChEMBL, quite diverse data
resources must be integrated.
• Most chemists are not used to advanced visual
analysis concepts and only have moderate confi-
dence in on-screen analysis so far. Visual repre-
sentations like heat maps and dendrograms are al-
ready used and intuitively understood, but combi-
nation in an integrated interactive environment is
not yet widespread. New interaction and analy-
sis concepts for the exploration of large chemical
databases need to be developed that are suitable
for chemists without expert knowledge in chem-
informatics and statistics.
2 SCAFFOLD HUNTER
Scaffold Hunter addresses the above mentioned chal-
lenges by means of a flexible framework for the in-
tegration of data sources and several interconnected
visual analysis components described in Sec. 2.1.
There are several workflows along the drug dis-
covery process that are related, but require slightly
different views on the data. Often, an overview on
the database contents is needed, both for evaluation
and for comparison. Applications include visualiza-
tion of several data sets at the same time, for instance
comparison of results from several assays, or data sets
stemming from multiple databases to rate their over-
lap or coverage of chemical space. An internal and
a commercial database could be compared to gauge
to what extent purchasing would increase the cover-
age of promising regions of chemical space, or where
patent issues might be relevant. It should be noted
that the visualization of this space is not restricted
to show what is contained in the database, but also
indicates gaps in the structural coverage, which give
hints on structurally simpler but still biologically ac-
tive molecules for synthesis or purchase.
A further task is the search for biologically ac-
tive molecules that may be promising for synthesis
to check suitability as potential drugs. Here, spots of
large potential biological activity have to be identi-
fied. Note that biological activity for the largest part
of the chemical space is not known, as the molecules
are not tested or not even synthesized, but can only
be derived indirectly, e.g., from the values of simi-
lar molecules with known activity. In addition, there
are also many other required or desired properties, as
for example synthesizability or bio-availability, which
need to be estimated, and are often approximated best
by experienced chemists with the help of computa-
tional analysis methods. Hence, the dynamic gener-
ation of new hypotheses and the integration of addi-
tional experimental data during the discovery process
requires a complex interplay between interactive vi-
sualization, analytical reasoning, computational anal-
ysis, and experimental evaluation and validation as il-
lustrated in Figure 1. A recent work-flow based on
the scaffold tree classification combines scaffolds that
are not annotated with bioactivity with scaffolds of re-
lated small molecules with known bioactivity and tar-
gets (Wetzel et al., 2010). The merging of the corre-
sponding trees allows to prospectively assign bioac-
tivity and to identify possible target candidates for
non-annotated molecules.
Most of the use-cases include exploration of the
chemical space, and therefore the core concept of
Scaffold Hunter builds upon a corresponding navi-
gation paradigm for orientation as described in Sec-
tion 2.1.1. As many use-cases also rely on the import
of data from heterogenous sources, Scaffold Hunter
provides a flexible data integration concept, which is
described in Sec. 2.3.
IVAPP 2012 - International Conference on Information Visualization Theory and Applications
628
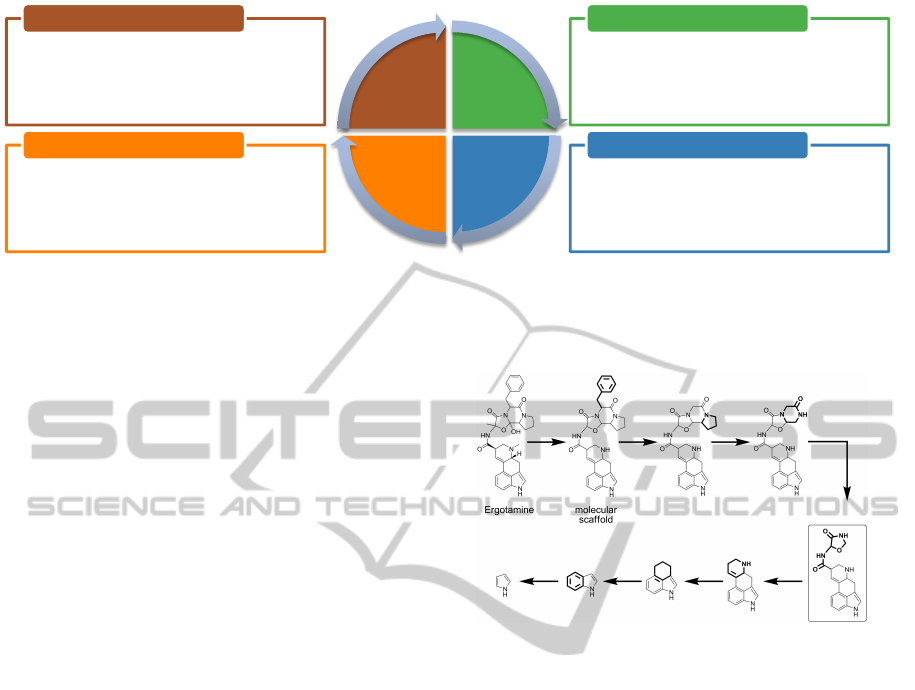
• The drug discovery process is a task that is not
suitable for a fully automatic process.
• The knowledge discovery process essentially
depends on the expertise of domain specialists, but
can greatly benefit from computational methods.
Analytical Reasoning
• In order to take advantage of publicly available
information quite diverse data resources must be
integrated.
• Experimentally obtained data can be investigated
to test hypotheses.
Data Integration
• Visualization of raw data as well as analysis results
requires different views that must be well
coordinated and linked in an intuitive manner.
• Views should be customizable to adapt to the
user’s needs and foster interactive exploration.
Interactive Visualization
• Statistical methods like clustering and classification
reveal relationships and patterns within the data.
• Approaches to organize chemical space should
take structural relations of compounds into account
to support revealing structure-activity relationships.
Automated Analysis
Analytical
Reasoning
Data
Integration
Automated
Analysis
Interactive
Visualization
Figure 1: Interactive visual analysis of the correlation between chemical structure and biological activity. The knowledge
discovery process is a cyclic procedure: Analyzing known data may allow to generate new hypotheses which lead to further
experiments. Results obtained here are again integrated into the tool for further investigation.
2.1 Visual Analysis Components
Since the search for drug candidates involves a com-
plex knowledge discovery process there is no single
best technique that reveals all relations and informa-
tion that might be of interest to the chemists. Scaf-
fold Hunter combines different approaches to catego-
rize and organize the chemical space occupied by the
molecules of a given compound set allowing the user
to view the data from different perspectives. Two im-
portant aspects here are structural features and prop-
erties of compounds. Relating structural character-
istics to properties like a specific biological activ-
ity is an important step in the drug discovery pro-
cess. Therefore, Scaffold Hunter supports to analyze
high-dimensional molecular properties by means of a
molecular spreadsheet and a scatter plot module. De-
veloping meaningful structural classification concepts
is a highly challenging task and still subject of recent
research. Two orthogonal concepts have emerged:
Approaches based on unsupervised machine learn-
ing and rule-based classification techniques, which
both have their specific advantages (Schuffenhauer
and Varin, 2011). Therefore, Scaffold Hunter sup-
ports cluster analysis using structure-based similar-
ity measures, a typical machine learning based tech-
nique, as well as a rule-based approach based on scaf-
fold trees. Comprehensive linkage techniques foster
the interactive study of different perspectives of a data
set providing additional value compared to isolated
individual views.
2.1.1 Scaffold Tree
In order to organize chemical space and to reduce
the number of objects that have to be visualized, we
use the scaffold tree approach (Schuffenhauer et al.,
2007). This approach computes an abstraction of the
molecule structures that allows to represent sets of
Figure 2: Creation of a branch in the scaffold tree.
molecules by single representatives, so-called scaf-
folds, for navigation. A scaffold is obtained from a
molecule by pruning all terminal side chains. The
scaffold tree algorithm generates a unique tree hierar-
chy of scaffolds: In a step-by-step process, each scaf-
fold is reduced up to a single ring by cutting off parts
that are considered less important for biological ac-
tivity, see Figure 2. In each step a less characteristic
ring is selected for removal by a set of deterministic
rules, such that the residual structure, which becomes
the parent scaffold, remains connected. By this means
the decomposition process determines a hierarchy of
scaffolds. As, depending on the task at hand, differing
aspects may be crucial to define relevant relations be-
tween scaffolds, the user can customize the rules for
scaffold tree generation. The resulting set of trees is
combined at a virtual root to a single tree which can
be visualized using graph layout techniques.
Each scaffold represents a set of molecules that
are similar in the sense that they share a common
molecular framework. Experimental results show
that these molecules also share common biologi-
cal properties, making the classification suitable for
the identification of previously unknown bioactive
molecules (Schuffenhauer et al., 2007). Furthermore
SCAFFOLD HUNTER - Visual Analysis of Chemical Compound Databases
629
the edges of the scaffold tree provide meaningful
chemical relations along which such properties are
preserved up to a certain extent and are therefore ap-
propriate for navigation (Bon and Waldmann, 2010).
Compounds in a chemical database will not com-
pletely cover the chemical space spanned by the cre-
ated scaffolds. Scaffolds that are not a representative
of molecules, but solely created during the scaffold
tree reduction step, are nonetheless inserted into the
tree. These virtual scaffolds represent ‘holes’ in the
database and may be of particular interest as a starting
point for subsequent synthesis. They represent pre-
viously unexamined molecules that may for example
exhibit higher potency.
Since the generation of a scaffold tree for a large
data set is a time consuming task, Scaffold Hunter al-
lows to compute and permanently store scaffold trees
using the default rule set proposed in (Schuffenhauer
et al., 2007) or a customized rule set which can be
compiled by means of a graphical editor.
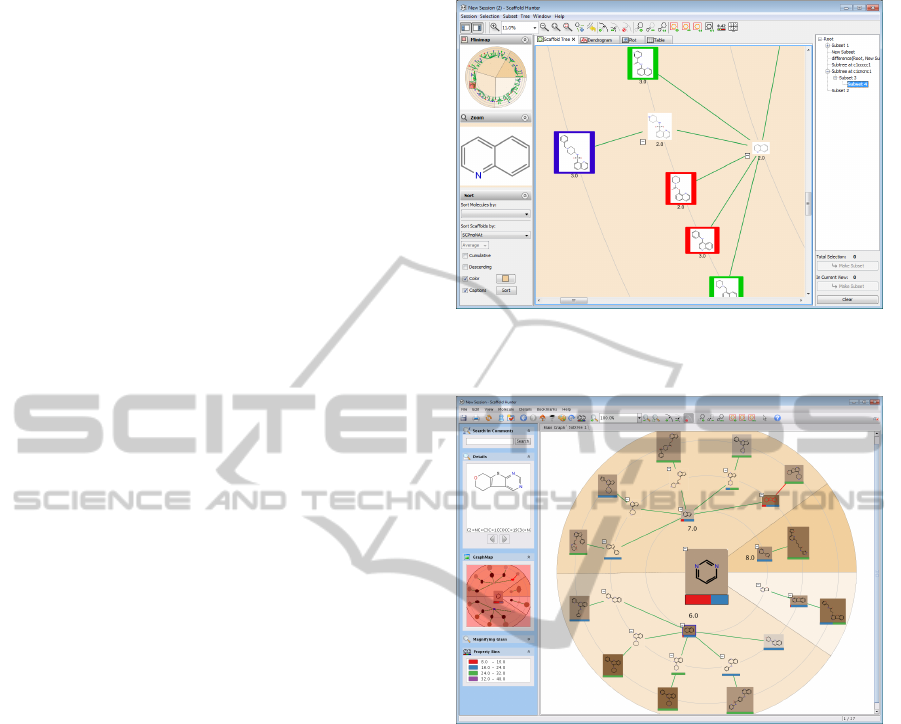
Scaffold Tree View. Based on the scaffold classi-
fication concept, Scaffold Hunter’s main view repre-
sents the scaffold tree. The implementation is based
on the toolkit Piccolo (Bederson et al., 2004) and sup-
ports to freely navigate in the scaffold tree view, as the
user interface allows grab-and-drag operations and
zooming. Zooming can be done either manually in
direction of the mouse cursor, or automatically when
the user switches between selected regions of interest.
The system then moves the viewport in an animation
to the new focus region, first zooming out automati-
cally to allow the user to gain orientation. At the new
focus region, the system zooms in again. For realiza-
tion of the Overview-plus-Detail concept, we imple-
mented a minimap. The minimap shows the whole
scaffold tree and the position of the viewport and al-
lows to keep orientation even at large zoom scales, see
Figure 3. Both the main view and the minimap allow
Pan-and-Zoom operations.
On startup, a user-defined number of levels is
shown, and an expand-and-collapse mechanism al-
lows the user to either remove unwanted subtrees
from the view or to explore deeper into subtrees of
interest. By default, the scaffold tree is laid out us-
ing a radial style and is always centered at the vir-
tual root. We decided not to allow the selection of a
new root for the following reason: As drug candidates
need to meet certain requirements regarding their bi-
ological activity and bio-availability, it will rarely be
necessary to explore trees over more than a few levels
(typically < 8). The molecules on deeper levels will
be too large and have too many rings to be relevant
for further consideration. However, in the case that
Figure 3: Close-up view of a scaffold tree, where properties
are represented by colored borders and text labels.
Figure 4: Layout of a subtree rooted at a scaffold of inter-
est with sorting and color shading. A sorting with respect
to a scaffold property can be applied to define the clock-
wise order of a scaffold tree, a background color shading of
segments reveals scaffolds with the same property value.
all molecules of the visualized subset share a com-
mon scaffold, the tree is centered on this scaffold and
the virtual root is hidden, as shown in Figure 4. Such
views allow to explore individual branches in detail.
In order to guide the chemist in his search for
a new drug candidate, scaffolds can be annotated
with property values derived from the associated
molecules, e.g. the average biological activity, or val-
ues directly related to the structure of the scaffold, e.g.
the number of aromatic rings. These properties can be
represented by several graphical features: The scaf-
fold and canvas background can be configured to in-
dicate associated categorical values by different col-
ors as well as continuous values by color intensity,
see Figures 3, 4. Edges can be configured to repre-
sent changes in property value by color gradient. Fur-
thermore, values can be mapped onto the size of a
IVAPP 2012 - International Conference on Information Visualization Theory and Applications
630

scaffold representation. Mapping property values to
graphical attributes allows both to get an overview on
the distribution of annotation values and to focus on
regions with specific values of interest. To show the
distribution of a selected molecule property for each
scaffold, property bins can be defined. A bar under
the respective scaffold image reflects the proportion
of molecules associated with the scaffold, that is as-
signed to a specified bin, see Figure 4. Property bins
may optionally indicate the values of the molecule
subset represented by a scaffold, or give the cumula-
tive values of the subtree rooted at the scaffold. This
information can help to select interesting subtrees for
deeper exploration.
The scaffold tree view provides a semantic zoom
that increases the level of graphical data annotations
with increasing zoom level, see Figure 5. Scaffolds
are represented using a 2D structure visualization,
which is sufficient for a good estimation of the chem-
ical behavior for the purpose of classification and the
investigation of potential drugs in an early stage. Dur-
ing navigation in zoom out mode, structure informa-
tion on scaffolds in the mouse pointer region is dis-
played in a magnifying glass window that can option-
ally be opened in the left side pane.
There are several requirements for layout meth-
ods within Scaffold Hunter which result from the
goals we defined for the application and also the ap-
proach taken. The layouts should represent the scaf-
fold tree hierarchy well, i.e., allow to easily follow the
bottom-up direction for navigation, to detect the scaf-
fold level, and to visually separate subtrees. In addi-
tion, the layout has to reflect a (circular) sorting of the
subtrees based on the user’s choice of a sorting scaf-
fold property. Also typical aesthetic criteria like edge
crossings and vertex-edge or vertex overlaps should
be taken into account. Several layout methods are im-
plemented, including radial, balloon, and tree layout.
All of them easily allow to satisfy our edge order, dis-
tance, crossing restriction, and vertex size constraints,
see Figures 4, 6. We give visual cues for the level affil-
iation of a scaffold by visualizing the radial circles as
thin background lines. In addition, we use a dynamic
distance between layers which is adapted according
to the zoom level. This allows to achieve good sepa-
ration of hierarchy levels and a clear depiction of the
tree structure in lower zoom levels, whereas in close-
up zooms scaffolds can still be represented together
with at least one child level.
2.1.2 Cluster Analysis
In cheminformatics cluster analysis based on molec-
ular similarity is widely applied since the 1980s
and can now be considered a well-established tech-
nique (Downs and Barnard, 2003) compared to the
novel scaffold tree concept. However, computing
an appropriate similarity coefficient of molecules is
far from trivial and many similarity measures have
been proposed (Maggiora and Shanmugasundaram,
2011). Common techniques to compare the struc-
ture of chemical compounds include their representa-
tion by bit vectors, so-called molecular fingerprints,
which encode the presence or absence of certain sub-
structures, and allow the application of well known
(dis)similarity measures like Euclidean distance or
Tanimoto coefficient. The choice of an adequate sim-
ilarity coefficient may depend on the specific task per-
formed or the characteristics of the molecules which
are subject to the analysis. To cope with the need for
various molecular descriptors Scaffold Hunter sup-
ports their computation by plugins.
We implemented a flexible clustering framework
including a generic interface which allows the user to
select arbitrary numerical properties of molecules and
to choose from a list of similarity coefficients. Fur-
thermore specific properties and similarity measures
for fingerprints and feature vectors are supported.
Scaffold Hunter includes a hierarchical clustering al-
gorithm and supports various methods to compute
inter-cluster similarities, so-called linkage strategies.
Dendrogram View. The process of hierarchical
clustering can be visualized by means of a dendro-
gram, a tree diagram representing the relation of
clusters. The dendrogram is presented as another
view and is supplemented by a modified spread-
sheet which can be faded in on-demand below the
dendrogram panel, see Figure 6. The spreadsheet
is tightly-coupled with the dendrogram: The order
of the molecules corresponds to the ordering of the
leaves of the dendrogram and an additional column
is added representing the cluster each molecule be-
longs to by its color. Scaffold Hunter fosters an in-
teractive refinement of clusters by means of a hori-
zontal bar which can be dragged to an arbitrary posi-
tion within the dendrogram. Each subtree below the
bar becomes a separate cluster. The spreadsheet dy-
namically adapts to the new partition defined by the
position of the bar.
When clustering large data sets dendrograms tend
to have a large horizontal expansion compared to the
vertical expansion. To take this into account we im-
plemented a zooming strategy that allows to scale
both dimensions independently giving the user the
possibility to focus on the area of interest. At higher
zoom levels the leaves of the dendrogram are depicted
by the structural formula of the molecules they repre-
sent. The sidebar contains a zoom widget that dis-
SCAFFOLD HUNTER - Visual Analysis of Chemical Compound Databases
631
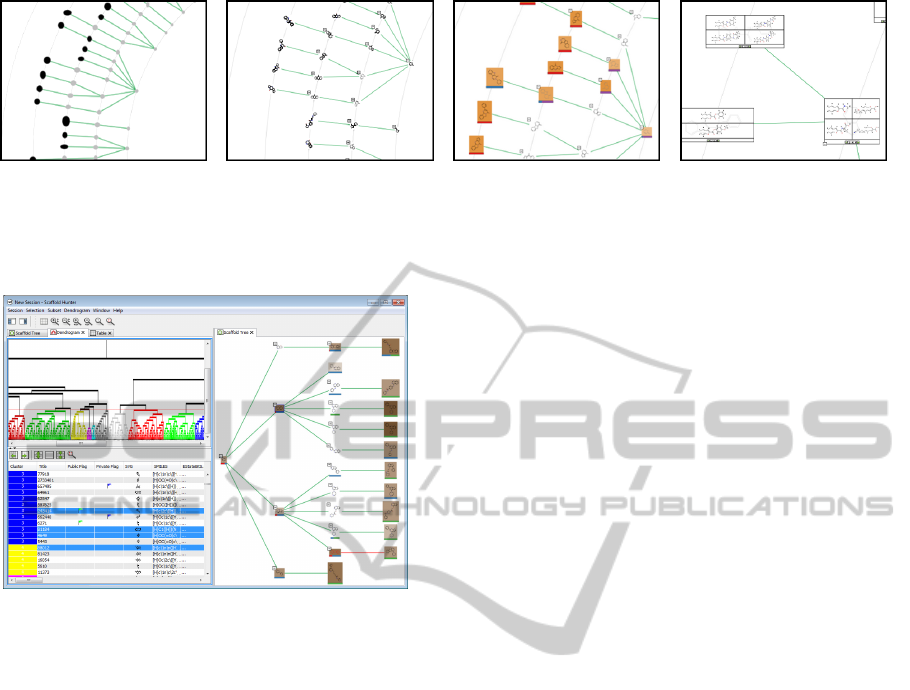
(a) Abstraction. (b) Structural formulas. (c) Full annotations. (d) Associated molecules.
Figure 5: Increasing level of detail with semantic zoom. Simplified representative shapes (a) are first replaced by structure
images (b), and finally the full set of currently selected data annotations is shown (c). Molecules associated with scaffolds (d)
can be displayed at lower zoom levels.
Figure 6: Split view showing a dendrogram combined with
a molecular spreadsheet (left) and a scaffold tree (right).
plays the molecule belonging to the leaf at the hori-
zontal position of the mouse pointer and is constantly
updated when the mouse pointer moves within the
dendrogram view. This allows the user to retain ori-
entation at lower zoom levels.
2.1.3 Molecular Spreadsheet
A molecular spreadsheet depicts a set of compounds
in table form, see Figure 6. Each row represents a
molecule and each column a molecular property. Our
implementation features an additional column show-
ing the structural formula of each molecule. The rows
of the table can be reordered according to the values
of a specified column, which allows the user, for ex-
ample, to sort the rows according to the biological ac-
tivity of the molecules and to inspect the molecules
successively, selecting or marking molecules of inter-
est. Deciding if a molecule is of interest for a spe-
cific task may, of course, depend on the expert knowl-
edge of the user who also wants to take different prop-
erties of the molecules into account. Therefore the
spreadsheet allows to freely reorder the columns and
to make the leftmost columns sticky. Sticky columns
always remain visible when scrolling in horizontal di-
rection, but are still affected by vertical scrolling. The
width and height of columns and rows, respectively,
is adjustable. Just like the scaffold tree view the side-
bar of the spreadsheet view features an overview map
and a detail zoom, showing the cell under the mouse
pointer in more detail. This is especially useful to in-
spect structural formulas that where scaled down to
fit into a cell or to completely view long texts that
were truncated to fit. The spreadsheet module is eas-
ily customizable and is reused as an enhancement of
the dendrogram view to which it can be linked.
2.1.4 Scatter Plot
Scaffold Hunter includes a scatter plot view that al-
lows for the analysis of multidimensional data. The
user can freely map numerical properties to the axes
of the plot and to various graphical attributes. At
least two properties must be mapped to the x- and
y-axis, respectively, but the user may optionally also
map a property to the z-axis turning the 2D plot into
a freely-rotatable 3D plot. In addition properties can
be mapped to the dot size or be represented by the
dot color, see Figure 8. This allows the user to visu-
ally explore the relationship of different properties, to
identify correlations, trends or patters as well as clus-
ters and outliers.
The sidebar contains several widgets showing ad-
ditional information or provide tools to interactively
manipulate the visualization of the data. When the
user hovers the mouse cursor over a data point, the
corresponding structural formula is shown in a de-
tail widget. The visible data points can be filtered
dynamically using range sliders and jitter can be in-
troduced to detect overlapping points. Selected or
marked molecules can be highlighted in the scatter
plot and single data points as well as regions can be
added to the selection.
2.2 Coordination and Linkage of Views
When multiple views of the data are provided, intu-
itive linking is of utmost importance for acceptance
IVAPP 2012 - International Conference on Information Visualization Theory and Applications
632

by chemists. Brushing and switching of views, e.g.,
from classification representations like dendrograms
to spreadsheets, are intuitive actions in the chemist’s
knowledge discovery process, and need to be sup-
ported in a way that allows to keep the orientation.
Scaffold Hunter incorporates several techniques af-
fecting all views in a similar manner.
Selection Concept. There is a global selection
mechanism for molecules, i.e. if a molecule is se-
lected in the spreadsheet view, for example, the same
molecule is also selected in all other views (Brush-
ing and Linking). All views support to select single
molecules or multiple at once by dragging the mouse
while holding the shift key. Since scaffolds represent
a set of molecules, not all of which must be selected
simultaneously, the coloring of scaffolds indicates if
all, none or only a subset is selected. If a scaffold
is selected, all associated molecules are added to the
selection. At a lower zoom level it is also possible
to select individual molecules, see Figure 5(d). Both,
the scaffold tree view and dendrogram view, are based
on a tree-like hierarchical classification. These views
also allow to select sets of related molecules belong-
ing to a specified subtree.
Subset Management and Filtering. In practice it
is not sufficient to just manage a single set of selected
scaffolds of interest. Therefore, Scaffold Hunter al-
lows to create and manage arbitrary subsets of the ini-
tial data set. The user can create a new subset contain-
ing all the molecules that are currently selected to per-
manently store the selection for later use. Of course,
it is possible to reset the selection to the molecules
of a stored subset. However, the subset concept is
much more powerful than suggested by this simple
use-case. Subsets can be created by means of a flexi-
ble filter mechanism based on rules regarding scaffold
and molecule properties deposited in the database, see
Figure 7. Filter rules can be stored and reapplied to
other molecule sets. A frequent task during the anal-
ysis of chemical compounds is the search for struc-
turally similar compounds and to filter large com-
pound databases by means of substructure search, i.e.
to create a subset consisting only of molecules that
contain a user-specified substructure. We have im-
plemented a fast graph-based substructure search ap-
proach (Klein et al., 2011) and integrated a structure
editor allowing to create search patterns graphically.
The result of a filtering can be highlighted in the cur-
rent view by setting the selection to the new subset.
All subsets created are presented at the right side-
bar in a tree-like fashion that reflects the relation of
subsets, see Figure 3. The user may perform the ba-
Figure 7: Filter dialog to define constraints.
sic set operations union, intersection and difference
on two or more sets leading to a new subset contain-
ing the result. Scaffold Hunter allows to create new
views showing only the molecules contained in the
selected subset. Furthermore the underlying subset of
the current view can be changed to a different subset
preserving the active mapping of properties to graph-
ical attributes.
The subset concept is suitable for the typical drill-
down approach in a chemical workflow, where the set
of considered molecules is reduced step by step. The
subset tree provides links back to upper levels of the
drill-down process to get back from dead ends and
fathomed areas of the chemical space under investiga-
tion. Restricting to subsets of medium-size helps the
user to preserve orientation and at the same time al-
lows for an efficient analysis and visualization. Even
though chemical databases may contain millions of
compounds, the interface capabilities are designed
and restricted to the visualization of dozens to only
several thousand compounds. However, the visual-
ization of all database entries as distinct entities at the
same time is hardly ever of interest for chemists.
Multiple Views and Connecting Elements. Scaf-
fold Hunter allows to inspect sets of molecules with
different views. Furthermore, it is possible to create
several views of the same type based on different sub-
sets. This is a prerequisite for the visual comparison
of different subsets, but requires techniques to help
the user to preserve orientation.
Scaffold Hunter supports labeling views to be able
to identify their source and how they were created,
e.g. by highlighting the underlying data set in the sub-
set tree. Each view comes with a specific toolbar and
sidebar (cf. Figure 3) and the GUI is adjusted when-
ever another view becomes active. However, for sev-
eral views the sidebar contains elements with a similar
intended purpose, but implemented in a view specific
manner. For example, all views offer a detail widget,
that works as a magnifying glass in the scaffold tree
view, as a zoom to the leaf node of the dendrogram,
shows a complete cell of a spreadsheet or details of
a dot in the scatter plot, respectively. A tooltip con-
SCAFFOLD HUNTER - Visual Analysis of Chemical Compound Databases
633
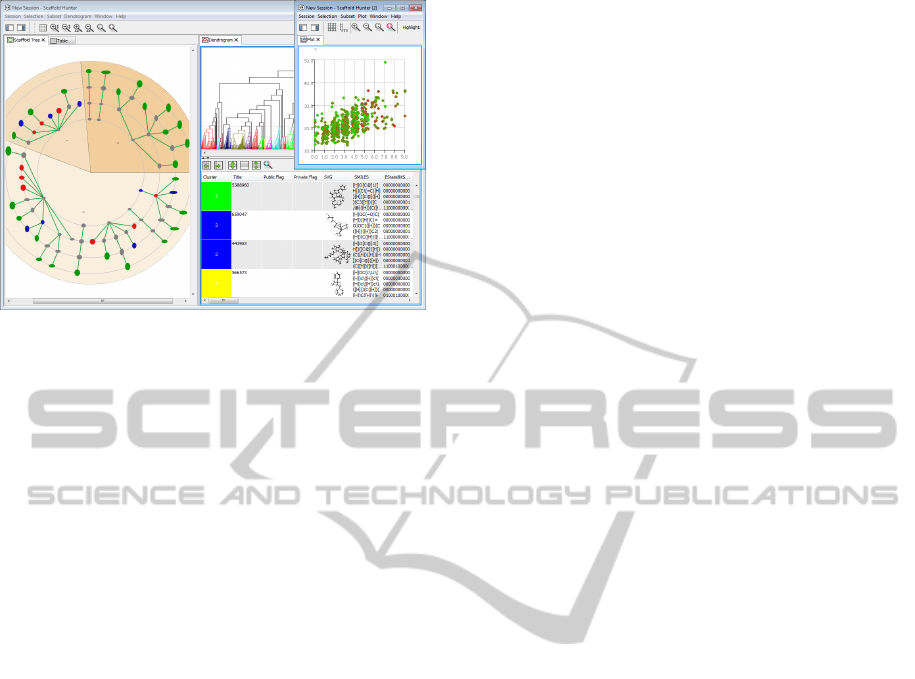
Figure 8: Several views showing data and statistical analy-
sis results complementary to the scaffold tree navigation.
taining a user-defined list of properties of a molecule
or scaffold as well as comments is consistently pre-
sented in several views. It is possible to annotate
specific molecules or scaffolds of interest and persis-
tently store comments, which can then also be viewed
by other users, if desired, to support joint work on a
project. In addition visual features like setting flags
to support orientation when moving back and forth
through several views are supported. Especially when
working with large molecule sets, it can be hard to re-
locate selected molecules in a different view. There-
fore all views support to focus the current selection,
e.g. by automated panning and zooming such that all
selected molecules are contained in the viewport.
Scaffold Hunter arranges multiple views by means
of a tabbed document interface, which most users are
familiar with and which allows to quickly switch be-
tween different views. To fully exploit the additional
benefit of different visual analysis components it is
important to consider multiple views at the same time.
Therefore, the tab pane can be split horizontally or
vertically and views can be moved from one tab pane
to the other, see Figure 6. Furthermore it is possible
to open additional main windows (cf. Figure 8) to
support work on multiple monitors.
Since the creation of subsets and the customiza-
tion of views is an important step in the knowledge
discovery process that should be preserved, the cur-
rent subset tree as well as the state of each view is
stored as a session and can be resumed later.
2.3 Data Integration
Chemical data on compounds is collected in different
databases that are accessible over web or program-
matical interfaces. The information stored as well as
the interfaces to retrieve them are highly heteroge-
neous. However, there are various standardized file
formats like structure data (SD) files which are com-
monly used and store sets of molecules with infor-
mation on their structure and their properties. Most
public databases support to export their content or
subsets, e.g. all compounds that where investigated
in the same bioassay, as SD file. Due to the sheer
amount of information and the need to prepare the
data to be accessible to our analysis techniques, we
rely on a data warehouse concept, i.e. compound
data can be extracted from different data sources, is
transformed, if necessary, and then loaded into a cen-
tral database once in a preprocessing step. Scaffold
Hunter only operates on this database. Compared to
a virtual database, where a unified view on different
databases is established by an on-line transformation
of queries and results, the data warehouse approach
allows to efficiently access data and to precompute
additional information, which is essential to facilitate
interactive analysis and navigation within the data.
Scaffold Hunter currently supports to integrate SD
files, CSV files and databases via customized SQL
queries. Since each data format is implemented as
a plugin, it is easily possible to add support for ad-
ditional data sources. The import framework allows
to define several import jobs that are processed subse-
quently. Since each imported data source may have a
different set of properties, defining an import job in-
cludes specifying a mapping to internal properties and
a merging strategy to cope with possible conflicts. It
is also possible to specify a transformation function
that can, e.g., be used to adjust the order of magni-
tude of the imported property values to the scale ex-
pected for the internal property. After an initial data
set has been stored, it is still possible to add additional
properties for each molecule. This allows to integrate
new experimental data at a later stage in the knowl-
edge discovery process, cf. Figure 1. In addition, it
is possible to calculate further properties that can be
derived from the structure of each molecule.
3 CONCLUSIONS & OUTLOOK
We presented Scaffold Hunter, a tool for the anal-
ysis of chemical space. There is already an active
user community that provides valuable feedback, and
the main concept of bioactivity guided navigation of
chemical space seems to be promising, which is also
backed by recent results (Bon and Waldmann, 2010).
Nonetheless the software could be extended by
features to address a broader community, with a
smooth integration into additional chemical work-
flows. Support for additional views and further analy-
IVAPP 2012 - International Conference on Information Visualization Theory and Applications
634

sis capabilities could help to boost the use of Scaffold
Hunter. The development and integration of addi-
tional functionality is encouraged by a modular soft-
ware architecture designed to be easily extendable
and by providing the software as open source.
A promising direction to enhance the currently
supported classification concepts based on tree-like
hierarchies is to support network-like structures. Re-
cently an extension of the scaffold tree approach was
proposed taking all possible parent scaffolds into ac-
count (Varin et al., 2011). This creates so-called scaf-
fold networks, which were shown to reveal additional
scaffolds having a desired biological property. Fur-
thermore networks can be used to represent struc-
tural similarities, e.g. derived from maximum com-
mon substructures, and might prove to be more flexi-
ble when ring-free molecules are considered or func-
tional side-chains should be taken into account. How-
ever, visualizing networks instead of tree-like hierar-
chies without compromising the orientation is chal-
lenging. New navigation concepts have to be devel-
oped and graph layout techniques must be customized
to the specific characteristics of such networks. We
plan to make use of the Open Graph Drawing Frame-
work (OGDF, 2011) for that purpose.
Due to the dynamic nature and the growing extent
of publicly available chemical data it might be help-
ful to also allow direct access to public resources from
within the GUI, e.g., by providing direct links to Pub-
Chem web pages for database compounds.
Scaffold Hunter is implemented in Java and freely
available under the terms of the GNU GPL v3 at
http://scaffoldhunter.sourceforge.net/.
ACKNOWLEDGEMENTS
We would like to thank the participants of student
project group PG552, the group of Prof. Waldmann,
in particular Claude Ostermann and Bj
¨
orn Over, Ste-
fan Mundt, Stefan Wetzel, and Steffen Renner for
their valuable suggestions and their contributions to
the project.
REFERENCES
Agrafiotis, D. K. and Wiener, J. J. M. (2010). Scaffold ex-
plorer: An interactive tool for organizing and mining
structure-activity data spanning multiple chemotypes.
Journal of Medicinal Chemistry, 53(13):5002–5011.
Bederson, B. B., Grosjean, J., and Meyer, J. (2004). Toolkit
design for interactive structured graphics. IEEE Trans.
Softw. Eng., 30(8):535–546.
Berthold, M. R., Cebron, N., Dill, F., Gabriel, T. R.,
K
¨
otter, T., Meinl, T., Ohl, P., Sieb, C., Thiel, K., and
Wiswedel, B. (2007). KNIME: The Konstanz Infor-
mation Miner. In Studies in Classification, Data Anal-
ysis, and Knowledge Organization (GfKL 2007).
Bon, R. and Waldmann, H. (2010). Bioactivity-guided
navigation of chemical space. Acc Chem Res.,
43(8):1103–14.
Downs, G. M. and Barnard, J. M. (2003). Clustering
Methods and Their Uses in Computational Chemistry,
pages 1–40. John Wiley & Sons, Inc.
Garcia-Serna, R., Ursu, O., Oprea, T. I., and Mestres, J.
(2010). iPHACE: integrative navigation in pharmaco-
logical space. Bioinformatics, 26(7):985–986.
Herhaus, C., Karch, O., Bremm, S., and Rippmann, F.
(2009). MolWind - mapping molecule spaces to
geospatial worlds. Chemistry Central Journal, 3:32.
IMI (2009). Innovative medicines initiative 2nd call, knowl-
edge management – open pharmacological space.
Irwin, J. J. (2009). Staring off into chemical space. Nat
Chem Biol, 5:536–537.
Klein, K., Kriege, N., and Mutzel, P. (2011). CT-Index:
Fingerprint-based graph indexing combining cycles
and trees. In IEEE 27th International Conference on
Data Engineering (ICDE), pages 1115 –1126.
Lounkine, E., Wawer, M., Wassermann, A. M., and
Bajorath, J. (2010). SARANEA: A freely avail-
able program to mine structure-activity and structure-
selectivity relationship information in compound data
sets. J. Chem. Inf. Model., 50(1):68–78.
Maggiora, G. M. and Shanmugasundaram, V. (2011).
Molecular similarity measures. Methods in Molecu-
lar Biology, 672:39–100.
OGDF (2011). The Open Graph Drawing Framework.
http://www.ogdf.net.
Schuffenhauer, A., Ertl, P., Roggo, S., Wetzel, S., Koch,
M. A., and Waldmann, H. (2007). The scaffold tree
- visualization of the scaffold universe by hierarchical
scaffold classification. J. Chem. Inf. Model., 47(1):47–
58.
Schuffenhauer, A. and Varin, T. (2011). Rule-based classi-
fication of chemical structures by scaffold. Molecular
Informatics, 30(8):646–664.
Varin, T., Schuffenhauer, A., Ertl, P., and Renner, S. (2011).
Mining for bioactive scaffolds with scaffold networks
- improved compound set enrichment from primary
screening data. J. Chem. Inf. Model.
Wetzel, S., Klein, K., Renner, S., Rauh, D., Oprea, T. I.,
Mutzel, P., and Waldmann, H. (2009). Interactive ex-
ploration of chemical space with scaffold hunter. Nat
Chem Biol, 5(8):581–583.
Wetzel, S., Wilk, W., Chammaa, S., Sperl, B., Roth, A. G.,
Yektaoglu, A., Renner, S., Berg, T., Arenz, C., Gian-
nis, A., Oprea, T. I., Rauh, D., Kaiser, M., and Wald-
mann, H. (2010). A scaffold-tree-merging strategy
for prospective bioactivity annotation of γ-pyrones.
Angew. Chem. Int. Ed., 49(21):3666–3670.
SCAFFOLD HUNTER - Visual Analysis of Chemical Compound Databases
635
