
String Searching in Referentially Compressed Genomes
Sebastian Wandelt and Ulf Leser
Humboldt-Universit
¨
at zu Berlin, Knowledge Management in Bioinformatics, Rudower Chaussee 25, 12489 Berlin, Germany
Keywords:
Genome Compression, Referential Compression, String Search.
Abstract:
Background: Improved sequencing techniques have led to large amounts of biological sequence data. One
of the challenges in managing sequence data is efficient storage. Recently, referential compression schemes,
storing only the differences between a to-be-compressed input and a known reference sequence, gained a lot
of interest in this field. However, so far sequences always have to be decompressed prior to an analysis. There
is a need for algorithms working on compressed data directly, avoiding costly decompression.
Summary: In our work, we address this problem by proposing an algorithm for exact string search over com-
pressed data. The algorithm works directly on referentially compressed genome sequences, without needing
an index for each genome and only using partial decompression.
Results: Our string search algorithm for referentially compressed genomes performs exact string matching for
large sets of genomes faster than using an index structure, e.g. suffix trees, for each genome, especially for
short queries. We think that this is an important step towards space and runtime efficient management of large
biological data sets.
1 INTRODUCTION
The development of novel high-throughput DNA se-
quencing techniques has led to an ever increasing
flood of data. Current techniques, usually summa-
rized under the term second generation sequencing
(SGS), are able to produce roughly the same amount
of data in about a week at a current cost of roughly
2000 USD. Is is predicted, that third generation se-
quencing deliver a further speed-up, reducing the
time and price for sequencing a human genome from
weeks to days and from thousands to under a hun-
dred USD, respectively (Schadt et al., 2010). As a
concrete example, the UK-based Wellcome Trust Se-
quencing Center recently reported that its throughput
in terms of DNA sequences has risen from 100KB/
day in the times of the human genome project to cur-
rently 1TB/day (Chiang et al., 2011).
A human genome consists of 23 chromosomes.
Each chromosome is a sequence of 50.000.000
and 250.000.000 nucleotides: A(denin), C(ytosin),
G(uanin), or T(hymin). In order to store a complete
genome of a human being, one needs more than 3 GB
(uncompressed). Sequence compression is one key
technology to cope with the increasing flood of DNA
sequences (Pennisi, 2011).
Substitutional or statistic compression schemes
can reduce the space requirements by up to 6:1 (one
base is encoded with up to 1.3 Bit) (Antoniou et al.,
2010; Pratas and Pinho, 2011). However, in many
projects only genomes from one species are consid-
ered. This means that projects often deal with hun-
dreds of highly similar genomes; for instance, two
randomly selected human genomes are identical to
an estimated 99.9%. This observation is exploited
by so-called referential compression schemes, which
only encode the differences of an input sequence with
respect to a pre-selected reference sequence. Using
space-efficient encoding of differences and clever al-
gorithms for finding long stretches of DNA without
differences, the best current referential compression
algorithm we are aware of reports a compression rates
of up to 500:1 (Deorowicz and Grabowski, 2011),
yielding 4 − 8 MB per genome.
In the future, many research facilities will need
to manage and analyze large sets of genomes. We
think that these genomes have to be referentially com-
pressed, in order to overcome the I/O- and storage
bottlenecks of todays computing systems in bioinfor-
matics(Kahn, 2011).
One important type of analysis is string search, i.e.
finding matching parts of the sequence with respect to
a given query. In this paper, we discuss string search-
ing on large sets of genomes G: Given an input query
95
Wandelt S. and Leser U..
String Searching in Referentially Compressed Genomes.
DOI: 10.5220/0004143400950102
In Proceedings of the International Conference on Knowledge Discovery and Information Retrieval (KDIR-2012), pages 95-102
ISBN: 978-989-8565-29-7
Copyright
c
2012 SCITEPRESS (Science and Technology Publications, Lda.)

Table 1: Rough storage requirements for approximate string search over 1000 genomes.
Element index-based index-less Referential search
Compressed genomes 6 GB 6 GB 6 GB
Index compressed genomes 3+ TB 0 GB 0 GB
Uncompressed reference sequence 0 GB 0 GB 3 GB
Index for reference sequence 0 GB 0 GB 7 GB
Sum 3+ TB 6 GB 16 GB
string q, the task is to find all matches of q in each
genome in G.
The traditional approach is to perform the search
on each genome in G using an index. However, since
these indices are often bigger than the uncompressed
input, this naive approach seems to be not feasible.
For instance, the raw data of 1000 uncompressed
genomes already needs 3 TB. Having an index struc-
ture, e.g. a compressed suffix tree, for each genome
will increase the data size further by factors of 3-5 or
more(Vlimki et al., 2009). Therefore, we think that
using one index structures for each referentially com-
pressed genome is not feasible in large-scale projects.
Another approach is to decompress all strings in
G, and perform index-less string search.
In this paper, we propose a third solution: search-
ing within compressed sequences. All input se-
quences were referentially compressed with respect
to a reference sequence, and for the compression one
usually needs an index structure for that reference.
Our idea is to use the existing index structure of the
reference for a string search algorithm on all com-
pressed genomes in G. Since all genomes in G were
referentially compressed with respect to the reference,
large substrings of the reference will occur in the
genomes in G as well, interrupted by SNPs and longer
variations. Therefore, our referential search algorithm
solves the string matching problem as follows: 1) It
finds all exact matches in the reference sequence us-
ing an index of the reference only and 2) it uses these
matches for identifying all exact matches in genomes
in G.
The space requirements for all three approaches
are compared in Table 1. We report on the run times
of different approaches in detail below.
The remaining part of the paper is structured as
follows. In Section 2, we discuss related work on
compression of genome sequences. We introduce a
general referential compression algorithm in Section
3 and propose a string search algorithm of referen-
tially compressed sequences in Section 4. A prelim-
inary evaluation is given in Section 5. The paper is
concluded with Section 6.
2 RELATED WORK
The increasing number of (re-)sequenced genomes
has lead to many compression algorithms. We only
review losless compression schemes here. In gen-
eral, these compression algorithms can be separated
into bit-manipulating, dictionary-based, statistical,
and referential approaches:
• Bit Manipulation algorithms exploit encodings
of two or more symbols into one byte (Vey, 2009;
Bharti et al., 2011; Mishra et al., 2010).
• Dictionary-based or substitutional algorithms re-
place long repeated substrings by references to a
dictionary built at runtime (Kuruppu et al., 2012;
Antoniou et al., 2010; Kaipa et al., 2010).
• Statistical or entropy encoding algorithms derive
a probabilistic model from the input. Based on
partial matches of subsets of the input, this model
predicts the next symbols in the sequence. High
compression rates are possible if the model al-
ways indicates high probabilities for the next sym-
bol, i.e. if the prediction is reliable(Duc Cao et al.,
2007; Pratas and Pinho, 2011).
• Referential or reference-based approaches are
similar to dictionary-based techniques, as they
replace long substrings with references. How-
ever, these references point to external sequences,
which are not part of the to-be-compressed input
data (Brandon et al., 2009; Kuruppu et al., 2010;
Pande and Matani, 2011; Grabowski and Deorow-
icz, 2011).
There is additional work on read compression, e.g.
(Bhola et al., 2011) and (Wan et al., 2011). The main
problem in read compression is the compression of
quality scores, e.g. (Chen et al., 2011). However, for
the compression of whole genome sequences, quality
scores are, so far, not important. This might change
in the near future.
There has been a lot of research focused on ef-
ficient string search in collections of string. Standard
techniques are Boyer-Moore(Boyer and Moore, 1977)
and the use of suffix trees(Ukkonen, 1995). There ex-
KDIR2012-InternationalConferenceonKnowledgeDiscoveryandInformationRetrieval
96

Figure 1: Example for relative compression.
ists also work on managing and searching genomic se-
quences in large databases, e.g. (Altschul et al., 1990;
Kent, 2002; Hunt et al., 2002). However, to the best of
our knowledge there exists no work on string search
over referentially compressed genomic sequences.
3 REFERENTIAL COMPRESSION
AND DECOMPRESSION
We denote strings with s,t. The length of a string s is
denoted with |s| and the substring starting at position i
with length n is denoted s(i,n). s(i) is an abbreviation
for s(i,1). All positions in a string are zero-based,
i.e. the first character is accessed by s(0). The con-
catenation of two strings s and t is denoted with s ◦t.
Although a genome can be encoded with four charac-
ters, i.e. A,C,G, and T, we allow arbitrary symbols.
For instance, symbol N is often used to indicate an
unknown base. Given two strings s and t, the longest
prefix-suffix match of s in t, is the longest string t
m
,
such that t = t
1
◦t
m
◦t
2
and s(0,|t
m
|) = t
m
.
In referential compression, one or more data se-
quences are compressed with respect to a reference
sequence, by only encoding differences between the
input and the reference. This yields loss-less com-
pression, i.e. based on the reference sequence and
the difference description it is possible to recover the
data sequences. A referential compression algorithm
needs to generate a set of referential matches with
respect to the reference. The output of our referen-
tial compression algorithm is a file of compression
blocks, such that each block is one of the following:
• Referential Block Re f B(i, j): The data sequence
matches the reference sequence at position i for j
characters
• Raw Block RawB(s): A string s is encoded raw
(for instance if there is no good matching refer-
ence block).
Consider the reference sequence:
re f erence = AGACATACCTACATAC
together with the data sequence
input = ACCTACACCCTAGACACC,
as shown in Figure 1. One compression of input with
respect to re f erence is
comp = (Re f B(6,7), RawB(CCCT ),Re f B(0,5),
RawB(CC)).
The first seven symbols of input match the sym-
bols 6 to 12 and are encoded as a referential block
Re f B(6, 7), followed by a raw block, RawB(CCCT ).
The raw block is followed by another referential
block, Re f B(0, 5), indicating that the input matches
the first six symbols of the reference sequence. The
last compression block is a raw block, RawB(CC).
Altogether, the input sequence is encoded with five
compression blocks.
The compression ratio is dominated by the syn-
tactical representation of referential blocks, especially
the efficient encoding of integer values, and the num-
ber and length of referential matches. In the remain-
ing part of the paper, we do not discuss efficient en-
codings of compression schemes further, since there
exists plenty of research on that topic already, for in-
stance (Brandon et al., 2009) and (Daily et al., 2010).
To efficiently find referential blocks, we use suffix
trees for the reference sequence. The suffix trees of
a reference sequence allows us to find longest prefix-
suffix matches of parts of the data sequences with re-
spect to the the reference genome.
In the following, we present our compression al-
gorithm for genome sequences in detail. Algorithms
do not show range checks for the sake of readability.
StringSearchinginReferentiallyCompressedGenomes
97

Algorithm 1 assumes one data sequence in Input (as a
string of symbols). The input string is traversed from
left to right, and depending on the current symbols
in the input and in the reference block, different sub-
routines are executed. The function FIND-MATCH
is used to find the longest prefix-suffix match of the
current input position with respect to the reference. If
the match is shorter than MIN symbols, or starts with
a non-base symbol, the function ENCODE-RAW is
used for raw encoding of the next input symbols. Oth-
erwise, the function ENCODE-REF is used for refer-
ential encoding of the next input symbols.
Algorithm 1: Compression Algorithm.
!th
1: P
in
← 0
2: P
raw
← 0
3: while P
in
<| In | do
4: (p,l)=FIND-MATCH
5: if l < MIN or Input(P
in
) /∈ {A,C, G,T } then
6: l =ENCODE-RAW(MIN-1)
7: else
8: Add RefB(p,l) to output
9: end if
10: P
in
= P
in
+ l
11: end while
Matches are required to have at least MIN charac-
ters, in order to avoid spurious matches. Our experi-
ments have shown that the mere length of genomes,
e.g. human genomes, causes a lot of unrelated
matches with less than 20-25 characters. Further-
more, the compression gain is very small for such
short matches, while finding them is expensive (each
of them needs one index-lookup). The encoding of a
raw sequence in Algorithm 2 is straight-forward: the
string Raws is filled with symbols from the input until
a normal base is found or a length-constraint is vio-
lated. In the end, Raw(Raws) is added to the output.
Algorithm 2: ENCODE-RAW(l) Function.
!th
1: Raws ← ””
2: while |Raws| < l or Input(P
in
) /∈ {A,C,G,T } do
3: Raws ← Raws ◦ Input[P
in
];
4: P
in
← P
in
+ 1;
5: end while
6: Add RawB(Raws) to output
We show the compression process for the com-
pression of (see Figure 1)
In = ACCTACACCCTAGACACC
with respect to the reference sequence
Re f = AGACATACCTACATAC.
Lets assume that MIN = 5. First, the longest
match for the start of In is looked up in Re f and
(6,7) is returned (for matching ACCTACA in the ref-
erence). Re f B(6,7) is added to the output. After-
wards the input position P
in
is 7, and the algorithm
tries to find a new match for CCCTA.... The longest
local match is (7,2), which is shorter than five, and
therefore RawB(CCCT ) is added to the output. After-
wards the input position P
in
is 11, and the algorithm
tries to find a new match for AGAC.... The result of
FIND-MATCH is (0,5), such that Re f B(0,5) is added
to the output. Next, the remaining string CC is added
as a raw block RawB(CC) to the output.
Decompression of the referentially encoded input
sequences is straightforward. Basically, all compres-
sion blocks are unfolded according to their definition
(raw or referential block). For decompression of data
sequences we do not need the compressed suffix trees
any longer, but only the reference sequence.
4 STRING SEARCH ALGORITHM
There exists plenty of work on string search algo-
rithms for (not compressed) strings. Usually, an in-
dex structure is computed, for instance, a suffix tree.
These index structures allow for fast access to sub-
strings.
However, in our case, it is not feasible to have
index structures over all compressed genome se-
quences. For the sake of efficiency and for minimiz-
ing storage requirements, we want to avoid comput-
ing these index structures (which are usually orders
of magnitudes bigger than the data sequences, see
Table 1). Thus, we have an index structure for the
reference sequence only. In the following, we show
how the index structure for the reference sequence can
be used to efficiently find matches over referentially
(w.r.t. that reference) compressed sequences.
Assume that we want to search for the query q =
ACC in the input Input from the example above. The
situation is depicted in Figure 2. Matches for the
query ACC can be inside a compression block or be
overlapping more than one block. More formally, if
a string s is a subset of another (referentially com-
pressed) string t, s must either:
1. be a substring of one referential block (actually
the dereferenced referential block),
2. be a substring of one raw block, or
3. be an string overlapping two or more compression
blocks.
Since this list contains all possible cases, we can
find all matches for a query q by solving each sub
KDIR2012-InternationalConferenceonKnowledgeDiscoveryandInformationRetrieval
98

Figure 2: Example for search of ACC.
problem. The first kind of substrings is easy to find,
since we have an index on the reference sequence.
The second kind of substrings can be found by search-
ing the raw blocks. The overlapping substrings can be
found by thoughtful investigation and partial decom-
pression of subsequences next to the beginning and
end of each block. All three steps are explained be-
low. The list of compression entries for our example
is
Entries = (Re f B(6,7), RawB(CCCT ),Re f B(0,5),
RawB(CC)).
The set of relative match entries is
RelMatchEntries = {Re f B(6,7), Re f B(0,5)}.
The set of raw entries is
RawEntries = {RawB(CCCT ),RawB(CC)}.
4.1 Finding Matches inside Referential
Blocks
Given the matches for q from the reference se-
quence, we can conclude matches inside relative
match entries immediately. Let Re f Matches =
{(i, j) | Re f erence(i, j) = q}, which can be easily
computed by using the suffix tree for Re f erence.
In order to find all positions of q occurring inside
referential blocks, we need to find all intervals in
Re f Matches which are included in at least one in-
terval of RelMatchEntries. In our example, the re-
sult would be the set {Re f B(6,7)}, since we have
Re f erence(6,3) = ACC and the string interval (6, 7)
includes the string interval (6,2).
Intervals could be naively computed by pairwise
comparison of each block in RelMatchEntries with
each block in Re f Matches. However, this is ex-
penisve, especially if we have many data sequences
and therefore many sets of RMEntries which need
to be checked against one Re f Matches set. A bet-
ter approach is to order the reference matches in
Re f Matches by position of the match, e.g. this yields
((0,5),(6,7)). This allows us to use binary search in
order to find matching subintervals. The complete al-
gorithm is sketched in Algorithm 3.
In our example, we have identified the block
Match(6,7) as the only referential block containing
Algorithm 3: Referential Blocks Algorithm.
1: Result ←
/
0
2: for all E ∈ RelMatchEntries do
3: Perfom binary search on sorted Re f Matches
for sub intervals of E, result is I
4: Result ← Result ∪ I
5: end for
the substring for ACC. The position of q inside a refer-
ential block can be obtained by subtracting the begin-
ning of the reference match (in our example 6) from
the position of the relative match entry (also 6, which
yields position 0).
4.2 Finding Matches inside Raw Blocks
The next class of potential matches are all complete
matches in raw blocks. By construction of our com-
pression algorithm, raw blocks usually
• are very short (less than MIN symbols) or
• contain only non-base symbols, e.g. N.
Therefore, direct substring search for occurrences
of q is efficient here. Since the query never contains
any N symbols, matches in raw blocks are easy to
find.
In our example, we have to check all raw blocks in
RawEntries for containing the query ACC, Neither of
the two raw entries, RawB(CCCT ) and RawB(CC),
contains ACC. Thus, after the second step we still
have yet only found one (out of the three) occurrences
of ACC in the input sequence.
4.3 Finding Overlapping Matches
The most difficult task is to find matches overlapping
two or more compression blocks. We partially de-
compress one substring for each overlapping area of
compression blocks. We apply the following steps for
each compression block B:
1. Decompress the last |q| − 1 characters of B into
string temp
2. Further decompress the following compression
entries until |temp| = 2 ∗ |q| − 2
StringSearchinginReferentiallyCompressedGenomes
99
The rationale is that an exact match starting in
B must match 1 ≤ x ≤ |q| − 1 symbols in B and
then |q| − x symbols in the next compression blocks.
Therefore, it is sufficient to partially decompress only
2 ∗ |q| − 2 symbols in totel. If the partially decom-
pressed string temp contains q, then the compressed
input sequence contains q as well. The position can
be easily computed from the match inside the string
temp and the position of the first compression block.
In our example, we have four compression entries
and need to decompress three overlapping strings:
• Overlap after Re f B(6,7): CA ◦CC = CACC,
• Overlap after RawB(CCCT ): CT ◦ AG = CTAG,
• Overlap after Re f B(0,5): CA ◦CC = CACC.
The string CACC contains the query ACC and
therefore we have found two additional matches (one
starting at the end of the first compression block
and one starting at the end of the third compression
block). The matching position inside the input can be
computed from the matching position inside the over-
lap string, yielding 6 and 15.
Combining the above results we obtain the fol-
lowing three positions as matches for the query ACC:
{0,6,15}.
5 EVALUATION
In the following section, we evaluate our proposed
compression and string search using different set-
tings. All experiments have been run on an Acer As-
pire 5950G with 16 GB RAM and eight Intel Core
i7-2670QM, on Fedora 16 (64-Bit, Linux kernel 3.1).
All size measures are in byte, e.g. 1 MB means
1,000,000 bytes.
As test data we have used HG19H(Kent et al.,
2002) as a reference and the Korean genome(Ahn
et al., 2009) as input. The Korean genome contains,
besides bases and N-symbols additional characters for
indicating base probabilities. In related work it is
common practice to replace all occurrences of these
additional characters by N, which yields lossy com-
pression. However, we have chosen to encode the
original sequence, in order to obtain lossless com-
pression. Our compression rate could be further im-
proved, if these additional symbols were replaced
by N. The compression time for a complete human
genome was 29 seconds. To the best of our knowl-
edge this is almost one order of magnitude faster than
the best existing approach for referential compres-
sion. Since our algorithms can be easily parallelized,
we think that the performance of our simple compres-
sion algorithm can be improved further.
Figure 3: Compression ratio.
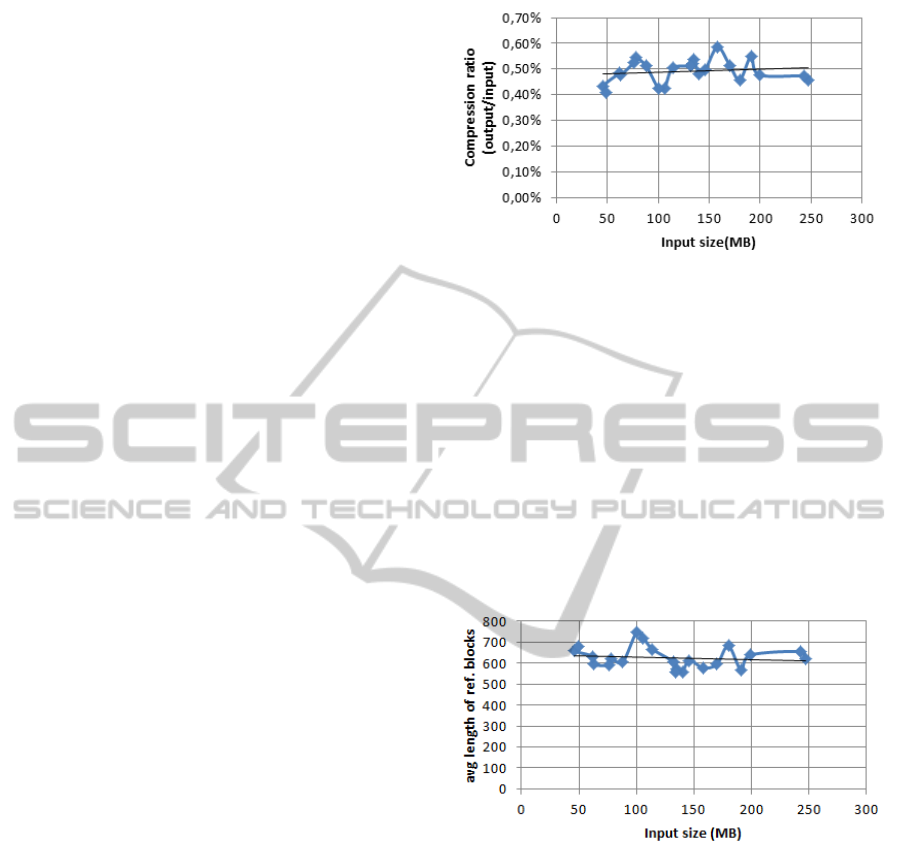
In Figure 3, the compression ratio is shown for
several data sequences (different chromosomes). The
compression ratio (size of the compressed output di-
vided by the size of the input) is roughly indepen-
dent of the size of the input, and is around 0.50 per-
cent. This means that we achieve a 20-fold com-
pression. The average length of referential blocks
is shown in Figure 4. It can be seen that the aver-
age length of referential blocks is also roughly inde-
pendent of the input chromosome and in average at
630 symbols/referential block. This coincides with
the background knowledge that around 99.9 percent
of two human genomes are equal.
Figure 4: Average length of ref. blocks.
Further analysis of the compression blocks
showed that the average length of raw blocks is 17
symbols. 54 percent of the blocks in a compressed
file are referential blocks and the remaining 46 per-
cent are raw blocks.
In order to evaluate our string search algorithm,
we have measured the run time for string search for
several queries. The result for a query string of length
12 is shown in Figure 5. The time needed for string
search grows linear with the length of the input se-
quence. This is due to the (linear) traversal of all com-
pression blocks during the search phase.
We ran experiments with different query lengths:
6, 12, 24, 48, and 96. Longer queries seem to slow
down the string search a little bit (order of few ms
comparing length 12 with length 96). Our investi-
KDIR2012-InternationalConferenceonKnowledgeDiscoveryandInformationRetrieval
100
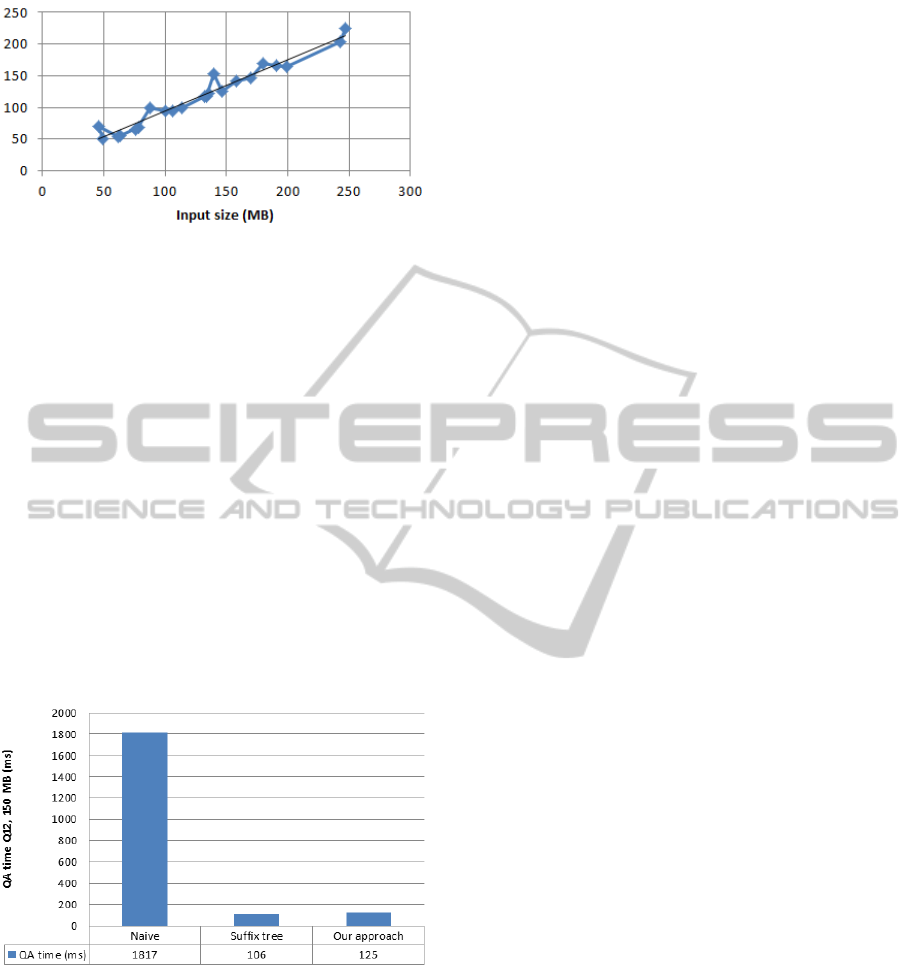
Figure 5: Answering times for Q12.
gation show that this is caused by the implementa-
tion of compressed suffix trees we have used: for
longer queries, the library spends more time to lookup
matches in the reference sequence.
Since, to the best of our knowledge, there ex-
ists no related work on string search over referen-
tially compressed genome sequences, we have imple-
mented the following competitors ourself for evalua-
tion purposes:
1. Naive search: First we completely decompress
the compressed input sequence into main mem-
ory and then search in-memory without an in-
dex, using bit-parallel string matching (Peltola
and Tarhio, 2003).
2. Index-based search: We use an existing suffix tree
for the uncompressed input sequence, in order
to find string matches.
Figure 6: Answering times for Q12.
The results are shown in Figure 6. It can be seen
that the naive approach is the slowest one, using al-
most 2 seconds to lookup the test string in the input
chromosome (150 MB). Therefore, we think that de-
compression is no solution for string matching with
respect to compressed files. The approach using suf-
fix trees is superior to our new approach, taking 106
ms compared to 125 ms. However, there are concerns:
1. The suffix tree of the index has to be created be-
forehand/offline. The additional time, we mea-
sured 10-40 seconds per chromosome for our test
genome, should be taken into account.
2. The suffix tree uses a lot of space. We measured
roughly an increase of factor 2-4 for our test chro-
mosomes. This means, that the index structure is
even bigger than the original uncompressed file.
Since suffix trees, or alternative index structures,
have to be created for each(!) input sequence, we
think that the small time overhead of our proposed ap-
proach is clearly acceptable. No extra data structures
have to be computed and stored offline. All we use
is the compressed file and the (one) index structure of
the reference sequence.
6 CONCLUSIONS AND FUTURE
WORK
One of the challenges in managing sequence data is
efficient storage and retrieval over compressed data.
In this paper, we addressed this problem by propos-
ing an algorithm for string search, which works di-
rectly on referentially compressed genome sequences.
Our evaluation shows that we can achieve similar run
times as if we had an index structure for each com-
pressed sequence. The ability to search biological se-
quences directly in a compressed structure opens new
ways for managing data in research groups. For in-
stance, a main-memory genome database, where all
genomes can be hold in RAM.
One important open challenge is approximate
string searching over referentially compressed se-
quences. We think that our search scheme can be ex-
tended in order to find approximate matches as well.
Scientific workflows have gained increased inter-
est during the last years in biology. The integration
of referential compression and string searching into
these workflows is one further open challenge.
REFERENCES
Ahn, S.-M., Kim, T.-H., Lee, S., Kim, D., et al. (2009).
The first Korean genome sequence and analysis: Full
genome sequencing for a socio-ethnic group. Genome
Research, 19(9):1622–1629.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and
Lipman, D. J. (1990). Basic local alignment search
tool. Journal of molecular biology, 215(3):403–410.
Antoniou, D., Theodoridis, E., and Tsakalidis, A. (2010).
Compressing biological sequences using self adjust-
ing data structures. In Information Technology and
Applications in Biomedicine.
StringSearchinginReferentiallyCompressedGenomes
101

Bharti, R. K., Verma, A., and Singh, R. (2011). A biological
sequence compression based on cross chromosomal
similarities using variable length lut. International
Journal of Biometrics and Bioinformatics, 4:217–223.
Bhola, V., Bopardikar, A. S., Narayanan, R., Lee, K., and
Ahn, T. (2011). No-reference compression of genomic
data stored in fastq format. In BIBM, pages 147–150.
Boyer, R. S. and Moore, J. S. (1977). A fast string searching
algorithm. Commun. ACM, 20(10):762–772.
Brandon, M. C., Wallace, D. C., and Baldi, P. (2009). Data
structures and compression algorithms for genomic
sequence data. Bioinformatics, 25(14):1731–1738.
Chen, W., Lu, Y., Lai, F., Chien, Y., and Hwu, W. (2011).
Integrating human genome database into electronic
health record with sequence alignment and compres-
sion mechanism. J Med Syst.
Chiang, G.-T., Clapham, P., Qi, G., Sale, K., and Coates, G.
(2011). Implementing a genomic data management
system using iRODS in the Wellcome Trust Sanger
Institute. BMC Bioinformatics, 12(1):361+.
Daily, K., Rigor, P., Christley, S., Xie, X., and Baldi, P.
(2010). Data structures and compression algorithms
for high-throughput sequencing technologies. BMC
bioinformatics, 11(1):514+.
Deorowicz, S. and Grabowski, S. (2011). Robust Rela-
tive Compression of Genomes with Random Access.
Bioinformatics.
Duc Cao, M., Dix, T. I., Allison, L., and Mears, C. (2007).
A simple statistical algorithm for biological sequence
compression. In Proceedings of the 2007 Data Com-
pression Conference, pages 43–52, Washington, DC,
USA. IEEE Computer Society.
Grabowski, S. and Deorowicz, S. (2011). Engineering rela-
tive compression of genomes. CoRR, abs/1103.2351.
Hunt, E., Atkinson, M. P., and Irving, R. W. (2002).
Database indexing for large dna and protein sequence
collections. The VLDB Journal, 11(3):256–271.
Kahn, S. D. (2011). On the future of genomic data. Science,
331(6018):728–729.
Kaipa, K. K., Bopardikar, A. S., Abhilash, S., Venkatara-
man, P., Lee, K., Ahn, T., and Narayanan, R.
(2010). Algorithm for dna sequence compression
based on prediction of mismatch bases and repeat lo-
cation. In Bioinformatics and Biomedicine Workshops
(BIBMW).
Kent, W. J. (2002). BLATThe BLAST-Like Alignment
Tool. Genome Research, 12(4):656–664.
Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M.,
Pringle, T. H., Zahler, A. M., and Haussler, D. (2002).
The human genome browser at UCSC. Genome Res,
12(6):996–1006.
Kuruppu, S., Beresford-Smith, B., Conway, T., and Zobel,
J. (2012). Iterative dictionary construction for com-
pression of large dna data sets. IEEE/ACM Trans.
Comput. Biol. Bioinformatics, 9(1):137–149.
Kuruppu, S., Puglisi, S. J., and Zobel, J. (2010). Relative
lempel-ziv compression of genomes for large-scale
storage and retrieval. In Proceedings of the 17th inter-
national conference on String processing and infor-
mation retrieval, SPIRE’10, pages 201–206, Berlin,
Heidelberg. Springer-Verlag.
Mishra, K. N., Aaggarwal, D. A., Abdelhadi, D. E., and
Srivastava, D. P. C. (2010). An efficient horizontal
and vertical method for online dna sequence compres-
sion. International Journal of Computer Applications,
3(1):39–46. Published By Foundation of Computer
Science.
Pande, P. and Matani, D. (2011). Compressing the human
genome against a reference. Technical report, Stony
Brook University.
Peltola, H. and Tarhio, J. (2003). Alternative algorithms for
bit-parallel string matching. In SPIRE, pages 80–94.
Pennisi, E. (2011). Will Computers Crash Genomics? Sci-
ence, 331(6018):666–668.
Pratas, D. and Pinho, A. J. (2011). Compressing the hu-
man genome using exclusively markov models. In
Rocha, M. P., Rodrguez, J. M. C., Fdez-Riverola, F.,
and Valencia, A., editors, PACBB, volume 93 of Ad-
vances in Intelligent and Soft Computing, pages 213–
220. Springer.
Schadt, E. E., Turner, S., and Kasarskis, A. (2010). A win-
dow into third-generation sequencing. Human molec-
ular genetics, 19(R2):R227–R240.
Ukkonen, E. (1995). On-Line Construction of Suffix Trees.
Algorithmica, 14(3):249–260.
Vey, G. (2009). Differential direct coding: a compression
algorithm for nucleotide sequence data. The Journal
of Biological Database and Curation, 2009.
Vlimki, N., Mkinen, V., Gerlach, W., and Dixit, K. (2009).
Engineering a compressed suffix tree implementation.
ACM Journal of Experimental Algorithmics, 14.
Wan, R., Anh, V. N., and Asai, K. (2011). Transforma-
tions for the compression of fastq quality scores of
next generation sequencing data. Bioinformatics.
KDIR2012-InternationalConferenceonKnowledgeDiscoveryandInformationRetrieval
102
