
A Roadmap to Implement a Quality Management System
Derek Flood, Fergal Mc Caffery and Valentine Casey
Dundalk Institute of Technology, Dublin Rd, Dundalk, Ireland
Keywords: Medical Devices, Software Process Improvement, Roadmaps, Quality Management System.
Abstract: In recent years the proportion and complexity of software in medical devices has increased considerably.
This has presented an opportunity for software development organisations to expand into the medical device
domain. Due to the high level of risk associated with medical devices, strict regulations must be adhered to
in order to market such products. One key aspect of these regulations is the necessity to have in place a
Quality Management System to help ensure an organisations’ ability to consistently meet customer and
regulatory requirements. This paper presents a roadmap which can be used to assist organisations, wishing
to develop medical device software to implement a Quality Management System.
1 INTRODUCTION
Advancements in technology have allowed medical
practitioners to provide a higher level of care to
patients and offer a wider range of treatment options.
However, when technology is used there is a risk to
the patient if that device should fail. To offset these
risks organisations must comply with the regulatory
requirements of the country where the device is to be
sold (Burton et al., 2006).
Software is becoming a more pivotal component
of medical devices due to its flexibility and its
ability to allow complex changes to be made,
without the need for hardware changes (Lee et al.,
2006), resulting in increased medical device
software complexity (Rakitin, 2006). In addition
software can now in its own right also be considered
a medical device (Mc Hugh et al., 2011). Therefore
software development organisations are subjected to
the same regulatory requirements as other medical
device organisations.
Software organisations now have an opportunity
to expand into the medical domain. These
organisations however must develop more stringent
processes in order to meet regulatory compliance. A
significant difficulty for such organisations is that
there is no prescribed method for performing
regulatory compliant software development
activities (McCaffery et al., 2010). Regulatory
bodies instead provide guidance documents
outlining what activities should be performed
(McCaffery et al., 2010).
To help address this shortfall this paper details a
software development process roadmap which can
be used to guide organisations through the
implementation of a Quality Management System
(QMS) which is compliant with the ISO 13485 (ISO,
2003) standard. The roadmap presented here has
been developed using the specific practices defined
in the Medi SPICE process assessment model, which
is designed to assess an organisations ability to
develop medical device software.
This paper is structured as follows. Section 2
introduces the Medi SPICE (McCaffery et al., 2010)
framework. Section 3 Introduces ISO 13485 and
details a roadmap for implementing a QMS, while
Section 4 outlines how the roadmap will be
validated. Section 5 details some of the limitations
of the roadmap and how these will be addressed in
the future. Conclusions are provided in Section 6.
2 MEDI SPICE
As regulatory bodies only detail what activities must
be performed, medical device organizations have
been compliance centric in their approach to
software development. As a result there has been
very limited adoption of software process
improvement within the medical device domain
(Denger et al., 2007).
Existing generic Software Process Improvement
(SPI) models, such as the Capability Maturity Model
Integration (CMMI
®
) (CMMI Product Team, 2006)
133
Flood D., Mc Caffery F. and Casey V..
A Roadmap to Implement a Quality Management System.
DOI: 10.5220/0004183601330138
In Proceedings of the International Conference on Health Informatics (HEALTHINF-2013), pages 133-138
ISBN: 978-989-8565-37-2
Copyright
c
2013 SCITEPRESS (Science and Technology Publications, Lda.)

and ISO 15504-5:2012 (ISO/IEC 15504-5:2012,
2012) (SPICE), do not provide sufficient coverage to
achieve medical device regulatory compliance
(McCaffery and Dorling, 2010). To address this
issue a medical device specific SPI framework, titled
Medi SPICE, has been developed.
The objective of undertaking a Medi SPICE
assessment is to determine the state of a medical
device organisation’s software processes and
practices. This is in relation to the regulatory
requirements of the industry and best practice with
the goal of identifying areas for undertaking process
improvement (McCaffery and Dorling, 2010). It can
also be used as part of the supplier selection process
when an organisation wishes to outsource or
offshore part or all of their medical device software
development to a third party or remote division
(Casey, 2010).
Medi SPICE is based upon the latest version of
ISO/IEC 15504-5:2012 and ISO/IEC 12207:2008
(ISO/IEC, 2008). It is being developed in line with
the requirements of ISO/IEC 15504-2:2003
(ISO/IEC, 2003) and contains a Process Reference
Model (PRM) and Process Assessment Model
(PAM). It also incorporates the requirements of the
relevant medical device regulations, standards,
technical reports and guidance documents.
The Medi SPICE PRM consists of 44 processes
and 15 subprocesses which are fundamental to the
development of regulatory compliant medical device
software. Each process has a clearly defined purpose
and outcomes that must be accomplished to achieve
that purpose.
Medi SPICE also contains a PAM which is
related to the PRM and forms the basis for collecting
evidence that may be used to provide a rating of
process capability. This is achieved by the provision
of a two-dimensional view of process capability. In
one dimension, it describes a set of process specific
practices that allow the achievement of the process
outcomes and purpose defined in the PRM; this is
termed the process dimension. In the second
dimension, the PAM describes capabilities that
relate to the process capability levels and process
attributes, this is termed the capability dimension.
3 QMS ROADMAP
The ISO 13485 – Medical devices - Quality
management systems – Requirements for regulatory
purposes, is an international standard defining the
requirements for the implementation of a QMS to be
used for the development of medical devices. The
standard not only covers software but also
incorporates hardware and related activities such as
production and servicing.
The main focus of the QMS is to help insure that
high quality processes are implemented and
monitored. The standard places a strong emphasis on
ensuring the organisation is committed to the quality
of their products, through effective process
management and a commitment to quality from all
levels of the organisation from top management
down.
To assist organisations to implement a QMS, a
roadmap has been developed. The roadmap is
divided into three phases. The first phase is project
planning and should occur at the start of a medical
device software development project.
The second phase is the system development
phase. During this phase the product is built using
the system development lifecycle.
The final phase is on-going and irregular
activities. The phase contains a number of practices
that should be performed during the development of
the product however; they do not fall under
individual stages of the software development
lifecycle. As these do not follow a strict sequence
they are not included as numbered steps.
Table 1 outlines the steps included within each
phase of the roadmap. The planning phase contains 6
steps, the systems development phase contains 16
steps and the on-going and event driven phase
contains 9 steps.
3.1 Project Planning Phase
The first section of the roadmap should be
performed prior to the development of the medical
device. During this phase the organisation will
establish the product scope and the procedures to be
used during the development of the medical device.
Step 1: Appoint a Quality Manager. The first step
in implementing a QMS is to assign a member of the
management team responsibility for overseeing and
reporting on the QMS, and promoting the awareness
of regulatory and customer requirements throughout
the organisation.
Step 2: Define Quality Objectives and Policy. The
quality objectives and policy outline the
organisations commitment to quality and guide the
processes used to ensure product quality. At this
stage the organisation’s quality manual
incorporating the scope of the QMS, and details and
justification for any exclusion and/or non-
application, is established.
HEALTHINF2013-InternationalConferenceonHealthInformatics
134
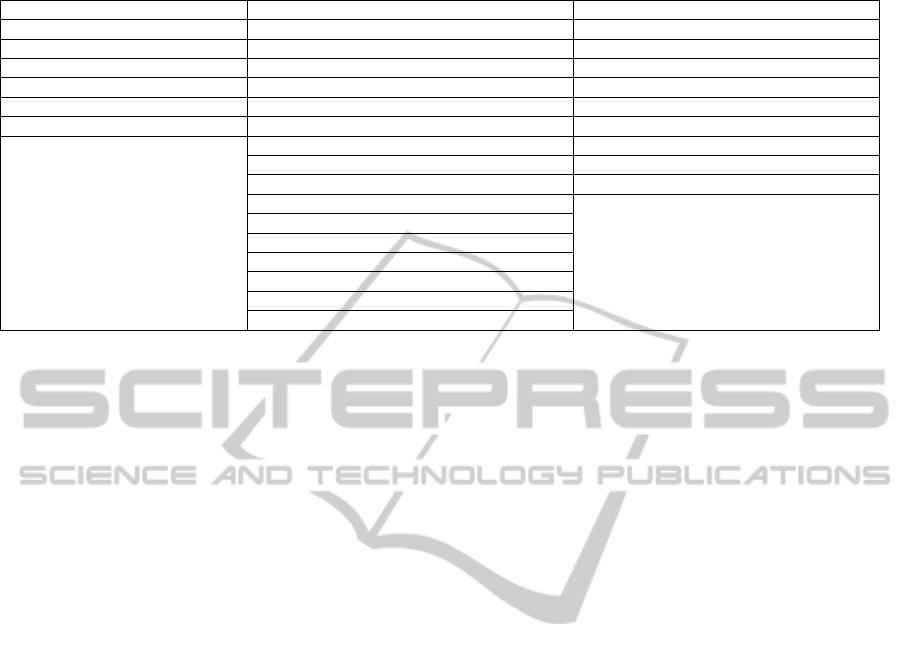
Table 1: Steps in the QMS roadmap.
Planning Phase Systems Development Phase On-going and Irregular Activities Phase
Appoint a quality manager Gather customer requirements Risk analysis
Define quality objectives Assign resources Document management
Establish product scope Analyse system requirements Software configuration management
Establish SDLC Design the system architecture Quality assurance
High level planning Software development Validation and verification
Low level planning Software requirements analysis Software review and audit
Software architectural design Problem resolution
Detail the software design Change request management
Software construction Acquisition
Software integration
Software qualification testing
Software installation
System integration
System qualification testing
Delivery
Customer support
Step 3: Establish the Product Scope. This step
involves the definition of the customer requirements
and an internal analysis of the organisations ability
to meet these. In addition to this a communications
interface should be established to ensure effective
communication between the customer and the
organisation.
Step 4: Establish the Software Development
Lifecycle (SDLC) for the Project. During this step
the organisation should define the software
development lifecycle that should be used during the
development of the product. This should include all
processes necessary for the QMS and the sequence
and interaction between these processes.
Step 5: High Level Planning. The next step is to
define the high level processes necessary for the
development of the product. For example,
development of a document management strategy.
Step 6: Low-level Planning. While high-level
processes define the overall strategy to be employed,
the low-level processes are focused on particular
areas or provide more detailed instructions to meet
the overall strategy.
3.2 System Development Phase
After the planning phase has been completed, the
organisation will begin to develop the medical
device. These steps follow the order of the software
development lifecycle and can be implemented in
the presented order.
Step 7: Gather Customer Requirements. The first
step of this phase is to determine the customer
requirements for the project. As part of this stage,
risk analysis should be performed and documented.
Step 8: Assign Resources. When the customer
requirements have been defined and understood top
management should ensure that adequate resources,
in terms of personnel and infrastructure, are in place
and documented.
Step 9: Analyse System Requirements. In this
step, non-functional requirements, potential risks
and performance expectations are identified. The
process, and consistency with customer
requirements, should be documented and
maintained.
Step 10: Design the System Architecture. If
external software components are to be used an
appropriate acquisition strategy should be in place.
The system architecture design should be consistent
with the system requirements and the process used
should be adequately documented.
Step 11: Software Development. The software
implementation process to be used during the project
should be documented and maintained. If any
deviation from this process is encountered the
relevant documentation should be updated.
Step 12: Software Requirements Analysis. The
requirements of the software component of a
medical device are analysed in the context of the
complete system and its operating environment.
Traceability and adequate testing should be ensured.
Risk control measures should be implemented and
any modifications to the system requirements should
be documented.
Step 13: Software Architectural Design. As part of
the QMS, it is necessary to ensure that the
architectural design is consistent with the system and
software requirements.
Step 14: Detail the Software Design. A detailed
software design document should be produced. This
shall be consistent with the system architecture. The
ARoadmaptoImplementaQualityManagementSystem
135

process used should be adequately documented.
Step 15: Software Construction. The software
units defined in the software design document
should be constructed. Detailed documentation,
including traceability, should be maintained as
required by the configuration management strategy.
Step 16: Software Integration. The software units
should be integrated into the complete software
system. The integrated system should be tested
independently from the testing of individual
components. The results from this testing, as well as
the strategy for integration and testing should be
documented and maintained.
Step 17: Software Qualification Testing. In this
step the integrated software system is tested. User
documentation should be updated as necessary. A
fundamental component of this stage is to ensure
that a regression test strategy is developed and
carried out. All processes used and results obtained
should be documented and maintained.
Step 18: Software Installation. Documented
procedures, including verification and acceptance
criteria, should be established as part of the QMS.
This documentation should be made available to
other agents who have been authorised by the
customer to perform installation activities.
Step 19: System Integration. The next step is to
integrate the software with the overall system. Each
component of the system should be independently
verified to ensure that they meet the system
requirements. The results of such verification and
the process used should be documented. Traceability
should be ensured.
Step 20: System Qualification Testing. As part of
the system qualification testing phase, a QMS
should ensure that the system tests can be traced
back to system requirements. The process used
should be documented and maintained.
Step 21: Delivery. The QMS requires that the
organisation should ensure that delivery conditions,
in accordance with the supply agreement, are met.
Step 22: Customer Support. The QMS should
ensure that the organisation monitors the operational
use and ensure that adequate support is available to
the customer. The organisation should also collect
and retain customer satisfaction data relating to both
the product and the customer service.
3.3 On-going and Irregular Activities
Phase
During the development process a number of
activities are performed at regular intervals or when
specific events occur. The schedule for these
activities should be defined during the planning
phase; however, the execution of these activities can
occur at any time.
Risk Analysis. At all stages of the product
development, the organisation should ensure that
adequate risk management activities are performed.
The level of risk analysis is dependent upon the class
of device that is being developed.
Document Management. Throughout the entire
development process a consistent document
management process should be in place. Each
document should be checked before distribution,
ensuring they are available where needed.
Software Configuration Management. During the
lifetime of the project, configuration items should be
maintained and accessible at the point of use. The
QMS should ensure that each configuration item is
uniquely identified and that a description of each
item is maintained.
Quality Assurance. The QMS dictates that regular
assessments should be performed in terms of both
the processes used during the development of the
product and the product itself. Through processes
assessment activities the organisation should
perform process improvement activities when
possible.
The organisation should also periodically assess the
achievement of the quality objectives and monitor
the performance of the QMS. When a deviation from
the quality objective is found, the organisation
should strive to take action as quickly as possible to
ensure that the quality of the product is not
negatively affected.
Part of the assessment and improvement
initiatives should be commitment from all relevant
departments within the organisation. This will
ensure assessment activities will not interfere with
the main functions of the organisational units under
assessment. The results of the assessment and any
process improvement activities that will take place
should be communicated to all stakeholders.
Validation and Verification. At regular intervals
the organisation should perform validation and
verification activities to ensure that the product is
correct and meets project requirements and quality
standards. The results of these activities should be
recorded and any problems identified should be
entered into a software problem resolution process.
The results of such activities should be
communicated to all relevant stakeholders.
Software Review and Audit. At regular intervals
the organisation shall prepare and conduct a
HEALTHINF2013-InternationalConferenceonHealthInformatics
136

software review and audit. If a product is found to be
non-conforming then the organisation should take
appropriate action either by removing the non-
conformities or by authorising its release under
concession. If the product is released under
concession, the organisation should ensure that it
still meets all regulatory requirements. The nature of
the non-conformity and the identity of the person(s)
authorising the concession should be documented.
Problem Resolution. The organisation should
ensure that any problem identified is recorded and
the root cause of the problem is diagnosed. The
organisation should have procedures in place to alert
users of the problem pending a fix or a change. A
solution for the problem should then be found and
implemented while tracking the status of the issue
until it is resolved. If no action is taken as a result of
a problem being identified, the organisation should
justify and record why no action has been taken. The
organisation shall maintain records of all customer
complaints.
Change Request Management. A documented
procedure should be in place to address change
requests. This procedure should ensure that an audit
trail is in place enabling traceability. Prior to the
implementation the organisation should assess the
impact of the change request on both the existing
system and other change requests. Additionally the
risks associated with the change request should be
analysed.
Upon implementation of the change request, the
organisation should perform all necessary
verification and validation activities. All
configuration items should be updated if impacted
by the change.
Acquisition. In the event the organisation shall
incorporate external software units in the product,
the organisation should ensure that an appropriate
acquisition process is followed. This process should
encompass a process for supplier tendering and
selection, acquisition of the product, acceptance
criteria and testing and a means for managing
changes and resolving issues with the supplier.
4 VA L I D AT I O N
Two aspects, order and completeness, of the
roadmap presented above will be validated. Order
refers to the placement of the practices in each step.
It will be necessary to ensure that all practices that
are depended upon are in place before an
organisation tries to implement any new practices.
The completeness of the roadmap refers to its ability
to cover the requirements of the ISO 13485 standard.
The order of the implementation steps will be
validated using the Medi SPICE dependency graphs
(Flood et al., 2012). The practices in each step will
be validated to ensure that all dependant practices of
each step are performed in the same or preceding
steps. If a practice is found to be performed in the
same step of the roadmap the organisation will be
alerted and instructed to perform this practice before
any dependant ones.
In order to validate the completeness of the
roadmap a number of qualified ISO 13485 assessors
will be asked to review it. A semi-structured
interview will then be conducted with the assessor.
5 LIMITATIONS AND FUTURE
WORK
The roadmap presented here is based on ISO 13485.
In the US the FDA produce a similar regulation,
FDA Regulation 21 CFR 820 (FDA, 2007), for
organisations wishing to distribute medical devices
within the US. Although both documents provide
similar guidance additional practices may be
required for medical device organisations wishing to
operate within the US. However, it should be noted
that the FDA are currently implementing a pilot
programme to accept a QMS that is compliant with
ISO 13485.
The next step of this work will be to perform a
similar analysis of the FDA Regulation 21 CFR 820.
The relevant best practices will be identified and any
additional requirements identified will be
incorporated into the existing roadmap to produce a
roadmap for organisations wishing to distribute
medical devices in Europe and the US.
The roadmap presented above allows an
organisation to create a QMS, assuming they have
no existing processes in place. In the case of
software development organisations wishing to enter
the medical device domain, some existing processes
may be in place and the roadmap will need to be
tailored for these organisations.
In addition to this there are some steps that may
not need to be performed, depending on the
circumstances of the organisation or product.
To address these issues an assessment method
that can be used to determine the organisations
existing processes and their specific requirements
will be developed.
ARoadmaptoImplementaQualityManagementSystem
137

6 CONCLUSIONS
This paper presents a software process roadmap for
the implementation of a QMS for organisations
operating within the medical device domain. In
order for medical device products to be sold, they
must be approved by the relevant regulatory body
within the region the device is to be sold, such as the
FDA in the US. The regulations set forth by these
organisations require that a QMS is in place during
the development and distribution of medical devices.
To assist organisations wishing to develop
medical devices, this paper details a software
process roadmap for implementing a QMS. This
roadmap is based on the ISO 13485, an International
standard detailing the requirements of a QMS for
organisations in the medical device domain, and the
base practices defined in the Medi-SPICE PAM.
ACKNOWLEDGEMENTS
This research is supported by the Science
Foundation Ireland (SFI) Stokes Lectureship
Programme, grant number 07/SK/I1299, the SFI
Principal Investigator Programme, grant number
08/IN.1/I2030 (the funding of this project was
awarded by Science Foundation Ireland under a co-
funding initiative by the Irish Government and
European Regional Development Fund), and
supported in part by Lero - the Irish Software
Engineering Research Centre (http://www.lero.ie)
grant 10/CE/I1855
REFERENCES
Burton, J., McCaffery, F., and Richardson, I., A risk
management capability model for use in medical
device companies. in International Workshop on
Software quality (WoSQ '06). 2006. Shanghai, China:
ACM.
Casey, V. (2010) Virtual Software Team Project
Management. Journal of the Brazilian Computer
Society, 16, 83 – 96.
CMMI Product Team (2006) Capability Maturity Model®
Integration for Development Version 1.2. Software
Engineering Institute, Pittsburch PA.
Denger, C., Feldmann, R., Host, M., Lindholm, C. &
Shull, F. (2007) A Snapshot of the State of Practice in
Software Development for Medical Devices. First
International Symposium on Empirical Software
Engineering and Measurement. Madrid, Spain.
FDA 2007. Title 21-Food and Drugs Chapter I --Food and
Drug Administration Department of Health and
Human Services subchapter h--Medical Devices part
820 Quality System Regulation. U.S. Department of
Health and Human Services.
Flood D., Mc Caffery, F., Casey, V., (2012)
“Understanding the Relationships Within the Medi
SPICE Framework”, The Seventh International
Conference on Software Engineering Advances
(ICSEA 2012).
ISO 13485:2003 (2003) Medical devices — Quality
management systems — Requirements for regulatory
purposes. Second ed. Geneva, Switzerland, ISO.
ISO/IEC 12207:2008 (2008) Systems and software
engineering - Software life cycle processes. Geneva,
Switzerland, ISO.
ISO/IEC 15504-2 (2003) - Software engineering —
Process assessment — Part 2: Performing an
assessment. 2003: Geneva, Switzerland.
ISO/IEC 15504-5:2012 (2012) Information technology -
Process Assessment - Part 5: An Exemplar Process
Assessment Model. Geneva, Switzerland, ISO.
Lee, I., Pappas, G., Cleaveland, R., Hatcliff, J., Krogh, B.,
Lee, P., Rubin, H. and Sha, L., High-Confidence
Medical Device Software And Systems. Computer,
2006. 39(4): p. 33 - 38.
McCaffery, F., Dorling, A. and Casey, V., (2010) Medi
SPICE: An Update. in International Conference on
Software Process Improvement and Capability
Determinations (SPICE). 2010. Pisa, Italy: Edizioni
ETS.
McCaffery, F. and Dorling, A., (2010) Medi SPICE
Development. Software Process Maintenance and
Evolution: Improvement and Practice Journal, 22, 255
– 268.
Mc Hugh, M., McCaffery, F. & Casey, V. 2011.
Standalone Software as an Active Medical Device In:
O'CONNOR ET AL (ed.) The 11th International
SPICE Conference Process Improvement and
Capability dEtermination. Dublin: Springer.
Rakitin, R., Coping with defective software in medical
devices. Computer, 2006. 39 (4): p. 40 - 45.
HEALTHINF2013-InternationalConferenceonHealthInformatics
138
