
New Algorithm for Analysis of Off-target Effects in siRNA Screens
Karol Kozak
1
, Sandra Kaestner
1
, Thomas Wild
2
, Andreas Vonderheit
1
, Benjamin Misselwitz
3
,
Ulrike Kutay
2
and Gabor Csucs
1
1
LMSC, ETH Zurich, Schafmattstr. 18, 8093 Zurich, Switzerland
2
Institute of Biochemsitry, ETH Zurich, Schafmattstr. 18, 8093 Zurich, Switzerland
3
Institute of Microbiology, ETH Zurich, Schafmattstr. 18, 8093 Zurich, Switzerland
Keywords: RNAi, siRNA, Off-target, Transcriptome, Bioinformatics, Cell based Screening.
Abstract: The occurrence of RNAi side effects called “off-target effects” is still a challenging aspect in the
interpretation of data from large-scale RNA interference screens. To reduce off-target effects, improved
algorithms have been developed for small interfering RNA (siRNA) design, but also chemical modifications
of double stranded RNA molecules were introduced by the various commercial providers. To aid the
analysis of large-scale screens, we present a new algorithm and tool for the prediction of potential off-target
effects that can be applied to RNAi experimental data. Our approach provides different possibilities to
search for homologies between individual siRNAs of cellular mRNAs. We demonstrate our approach on a
ribosomal RNAi screening dataset.
1 INTRODUCTION
RNA interference (RNAi) has become a powerful
method for post-transcriptional silencing of specific
genes (Hannon, 2002). RNAi comprises different
small RNA molecules, which all make use of the
RNA-induced silencing complex (RISC) in order to
knock-down proteins. The current paper
concentrates on siRNA (small inhibitory RNA) (see
Figure 1). These RNA molecules are double
stranded RNA of a length of 21 bp (basepairs). The
two strands are called antisense (active, guide) and
sense (inactive, star), whereas the antisense strand
will bind to the corresponding mRNA. The first 2 –
8 bases of the antisense siRNA is called seed region
and at bases 8 – 10 is the cleavage site.
For knock-down/screening purposes different
companies offer sets of siRNAs targeting the whole
genome (or a subset of it) for various organisms.
Typically, they offer at least three different siRNAs,
for each target gene. These siRNAs can be used
either as single siRNA or can be mixed and used as a
pool of siRNAs. In this paper we concentrate on
single siRNAs and don’t deal with the specific
issues connected to pooling. The main reason for
offering several siRNAs per target is the varying
knock-down efficiency of the individual oligos and
the occurrence of off-target effects (that can have a
number of biological reasons). In our study, we
focus on sequence-dependent off-target effects that
can be attributed to the binding of the siRNA to
other mRNAs than their target mRNA (Fedorov et
al., 2005); (Jackson et al., 2006); (Fedorov et al.,
2006). This effect is caused by a high degree of
sequence complementarity/similarity. The
specificity of the siRNA sequence is thus a crucial
factor in an RNAi experiment (Semizarov et al.,
2005). Gene expression silencing through the RNAi
machinery works perfectly if the siRNA is totally
complementarity to its target mRNA. Single
nucleotide mismatches between the siRNA and the
target mRNA decrease the rate of mRNA
degradation (Haley and Zamore, 2006); (Elbashir et
al., 2001). The algorithms of the different companies
for generating the best siRNA sequence typically
take this into account and check and exclude siRNA
sequences that have total complementarity to other
than the target mRNA. However, also partial
complementarity between siRNA and mRNA seems
to result in a silencing effect (Jackson et al., 2006).
Based on this tolerance, siRNAs could have up to
hundreds of potential target sequences in the
genome. Currently, the degree of complementarity
between the two sequences needed for silencing is
not well defined.
Sequence-dependent off-target effects can be
253
Kozak K., Kaestner S., Wild T., Vonderheit A., Misselwitz B., Kutay U. and Csucs G..
New Algorithm for Analysis of Off-target Effects in siRNA Screens.
DOI: 10.5220/0004185802530260
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2013), pages 253-260
ISBN: 978-989-8565-35-8
Copyright
c
2013 SCITEPRESS (Science and Technology Publications, Lda.)

caused by a number of mechanisms and they are
summarized in Table 1.
First of all, it has been reported that off-target
effects occur with a high probability, if the siRNA
shows ~90% complementarity (17 nucleotides out of
19) to an off-target gene (Birmingham et al., 2006);
(Jackson et al., 2003). However, a 21-nucleotide
double-stranded RNA sharing only partial
complementarity with an mRNA is still competent to
cause gene silencing via translational repression
(Saxena et al., 2003); (Jackson et al., 2003). It seems
that already as few as 11 contiguous
complementarity nucleotides or a total of 15 are
sufficient to reduce the level of mRNA transcripts
(Jackson et al. 2003). The complementarity of the
siRNA seed region (the first 2–8 bases of the
antisense siRNA-strand) plays a major role in the
occurrences of off-target effects (Jackson et al.,
2006) (see Figure 1). Further analyses showed a high
tolerance for mismatches outside of the seed region,
whereas differences within this 5’ end of the siRNA
are barely tolerated (Amarzguioui et al., 2007);
(Lewis et al., 2005); (Doench et al., 2003).
The center region of the siRNA is important to
stabilize the siRNA-mRNA-duplex and to enhance
mRNA degradation (Saxena et al., 2003). Alemán
and colleagues analysed this central region, which
comprises the cleavage site of the mRNA (position
8-10 of the antisense strand, see Figure 1). They
deciphered that mismatches in this region of the
siRNA seem to be critical (Alemán et al., 2007) and
result in no cleavage. Additionally, they also tested
the aspect of a G:U wobble and discovered that the
G:U base pair is recognized like an authentic
Watson-Crick base pair in the anti-sense RNA-
mRNA duplex. This wobble base pairing expands
the range of potential targets for a specific siRNA.
Figure 1: Structure of an siRNA: 21 bp RNA duplex with
2 nucleotide 3’ overhang on each strand, the two strands
are called antisense or active or guide strand and sense or
inactive or star strand, respectively, the first 2 – 8 bases of
the antisense strand are called seed region and at bases 8 –
10 of the antisense siRNA strand is the cleavage site.
Anderson et al., calculated the frequencies at
which the seed hexamers appear in the 3’UTR
transcriptome and called this the seed
complementarity frequency (SCF) (Anderson et al.,
2008). They discovered that siRNAs with a low SCF
also have a low probability of generating an off-
target effect. Finally, Ui-Tei et al. found that the
seed-dependent off-target effect is highly correlated
with the thermodynamic stability in the duplex
formed between the seed region of the siRNA guide
strand and its target mRNA (Ui-Tei et al., 2008).
Off-target effects seem to occur if there exists a high
thermodynamic stability in the 5’ region caused for
example through a high G/C content (Lin et al.,
2005). This leads to the conclusion that it is not the
absolute number of mismatches but probably the
overall stability of the siRNA-mRNA duplex what
determines the success of a silencing event.
In order to predict off-target effects, a number of
methods or algorithms can be applied. For example,
the Basic Local Alignment Search Tool (BLAST)
(Altschul et al., 1990) is adopted to find nearly exact
homologies. Although BLAST is an excellent tool
for broad sequence alignments, it falls short in its
ability to accurately predict small local homologies.
Other bioinformatics tools (Alemán et al., 2007; Lin
et al. 2005), which don’t have this shortcoming, try
to predict interactions between siRNAs and mRNAs.
But unfortunately, these sequence-based prediction
tools frequently don’t consider specific off-target
parameters like: target site location, 3’ UTR
conserved regions, design specificity. Here, we
describe a novel method and a software
environment, supporting the analysis of potential
off-target effects for every siRNA of interest. This
environment enables researchers to determine
potential off-target effects in high throughput siRNA
experimental results.
2 METHODS
2.1 Concept for a Bioinformatics
Analysis of Off-target Effects
Available sequence analysis tools fail to reliably
predict off-target effects for siRNA sequences.
Building upon current understanding for the
occurrence of off-target effects, a new modular
analytic process is introduced here. This process can
be specifically adapted to a variety of options in
results interpretation to identify potential off-target
genes for every siRNA of interest. In the next
subsection, different scenarios for a meaningful
application of such an off-target analysis are
elaborated.
To verify the results of an RNAi screen that
relied on e.g. 4 different siRNAs per target the
screening results are evaluated with respect to how
many of the 4 siRNA per target gave rise to a
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
254
phenotype. If all four siRNAs show an effect of
similar magnitude, one can reasonably be sure that
the downregulation of the intended target gene had
worked. However, most often, not all siRNAs for
one target yield a similar phenotype, and frequently,
the 4 siRNAs show a graded response in the
respective assay and sometimes only one or two
siRNAs show an effect, whereas the others don’t
show any effect. Very often 3 out 4 siRNAs are also
consider as a strong candidate. By performing the
off-target analysis, one can determine whether for
such siRNA there exists such a complementarity
with a top hit or not.
Table 1: Cause for sequence-dependent off-target effects.
1 Causes for off-target effects References
2 nearly exact complementarity
(Jackson et al., 2003;
Birmingham et al., 2006)
3
15 nucleotides (nt) in total or even 11
continuous nt match
(Jackson et al., 2003)
4 seed region complementarity
(Jackson et al., 2006;
Lin et al., 2005;
Amarzguioui et al., 2003)
5
miRNA function (seed region-3’UTR
conserved region complementarity)
(Doench et al. 2003)
6
multiple occurrences of the seed
region in an mRNA sequence
(Doench et al., 2003;
Lin et al., 2005)
7
complementary region at the
cleavage site, center of the siRNA
(Alemán et al., 2007;
Saxena, 2003)
8 tolerance of G:U wobble (Alemán et al., 2007)
9 seed complementation frequency (Amarzguioui et al., 2003)
10 high G/C content in the seed region (Lin et al., 2005)
2.2 The Analytic Process
Potential off-target effects are predicted based on
sequence complementarity regions between siRNAs
and mRNAs. For flexibility and extensibility
reasons, the process is composed as a set of steps,
which must be performed in sequence to get to an
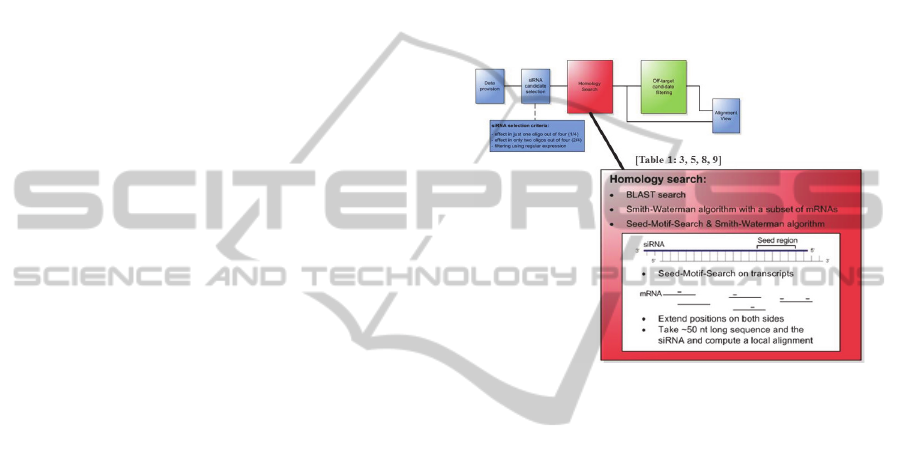
effective analysis (see Figure 2).
After selecting the potential off-target siRNAs,
the next step is to find homologies between these
siRNAs and all mRNAs. This concept contains
many variants for such a complementarity search
using different algorithms to perform a sequence
alignment between siRNA and mRNA. A detailed
description of the different complementarity search
strategies is given in the next subsection.
The resulting list of a complementarity search
can be too long to find the important results just by
visual inspection. Therefore the next step is to filter
this list to reduce its size to meaningful results (see
section filter options).
2.2.1 Complementarity Search
In this analysis step, it can be determined if there
exists a complementary region between the selected
siRNA sequences and the mRNAs. Many different
sequence alignment algorithms are available to
perform such a complementarity search, but they are
not optimal for the purpose of this process step by
default. Therefore, three different strategies for the
use of these algorithms have been developed to find
nearly exact complementary regions as well as small
local complementarities (see also Figure 2).
Figure 2: General structure of the concept for analysing
screening results of off-target effects. Three variants for a
complementarity search to find potential off-target effects,
which correspond to type of off-targets in Table1: lines 3,
5, 8, 9.
2.2.2 BLAST Search
The Basic Local Alignment Search Tool (BLAST)
(Altschul et al., 1990) is one of the most popular
algorithms for complementarity search and can be
applied to find nearly identical gene regions for a
specific siRNA sequence. Unfortunately, the
BLAST algorithm is not applicable to find all
potential off-target candidates, because genes with
only partial complementarity will be missing. A
complementarity between the seed region (positions
2-7 on the antisense strand) of the siRNA and the
mRNA sequence might be sufficient to cause off-
target effects. But initially, BLAST looks for
continuous matches that are at least 7 nt long and
thus would overlook genes with only a seed region
complementarity (Table 1: lines 2 and 3). Despite
this disadvantage, BLAST is an effective tool to find
out immediately if obvious off-target genes exist
with a nearly identical nucleotide sequence to the
siRNA (Table 1: line 1).
In contrast to BLAST, other local alignment
NewAlgorithmforAnalysisofOff-targetEffectsinsiRNAScreens
255

algorithms can find small partial complementarities
between siRNA and mRNA sequences. Therefore,
the developed concept offers, besides the BLAST
search, two different alternatives of building a local
alignment without getting into the runtime problem.
2.2.3 Smith-Waterman Algorithm
The Smith-Waterman algorithm is an accurate
algorithm used to build local alignments between
two sequences (Smith and Waterman, 1981). Since
its use with all mRNAs from the database is not
practicable, a feasible alternative is to limit the
number of mRNAs to approximately 200. The set of
200 mRNAs are mRNAs that have highest
alignment score with the siRNA. On a Windows 7
system (two Intel Xeon Quad-core 2.00 GHz CPUs,
16 GB RAM), the analysis of a 20 genes (4
oligonucleotides) library constructs took about 1
hour. By reducing the number of sequences it is
possible to perform a local alignment for all the
siRNAs.
2.2.4 Seed-Motif-Search Combined with the
Smith-Waterman Algorithm
Because of the mentioned runtime problem when
performing a local alignment with the Smith-
Waterman algorithm, a third variant to search for
complementarity is introduced here. In this variant,
an initial step reduces the length of the mRNA
sequences to enable the use of a local alignment
algorithm. This reduction is made because the seed
region of the siRNA seems to play a significant role
in causing off-target effects. At the beginning, all
occurrences of the seed motif of every siRNA are
localized in the genes (see Figure 3). After detecting
this small region, a sequence of ~50 nt around this
seed motif is cut out in the mRNA. Thus, as a result
of this first step, a huge number of sequences of ~50
nt in length are obtained containing the seed region
of each siRNA. Due to the small length of the
sequences it is now possible to perform a local
alignment with the Smith-Waterman algorithm.
2.2.5 Filter Options
The result of the complementarity search is a list of
potential off-target candidates. This list can be very
large including many false positives. To reduce its
size and to get only the most probable off-target
candidates, a great number of filtering options are
provided. The following filter options present a
central part of the analysis concept (see also Figure
3):
• Threshold filter: Only alignments that are higher
or lower than a specific threshold alignment score
• Position of matches in the alignment: The
positions at which the alignment should contain a
match can be specified with this filter. It can be
used, for example, to show only alignments
matching at position 9-11 in the siRNA sequence,
because this central region seems to play an
important role in the occurrence of off-target effects
(Table1:line 6). Optionally also a G:U wobble can
be tolerated as a match in the alignment (Table 1:
line 7).
• Number of matches in the alignment: The total
number of matches that should at least occur in the
alignment is defined in this filter (Table 1:line 8).
• Length of a continuous match in the alignment:
With this filter, the length of a continuous match can
be determined, e.g. the occurrence of a stretch of at
least 11 bases in the alignment (Table 1: line 2).
• Location within the mRNA: The location of the
alignment within the mRNA is specified in this
filter. For off-target it is important that siRNA
should match in the 3’UTR of the mRNA (Table 1:
line 3).
• Location within the 3’ UTR conserved region:
The conserved regions within 3’ UTRs of human
mRNAs are collected from UTRdb (Grillo et al.
2010).
• Multiple complementary regions of a siRNA in
one mRNA: Only the results of a siRNA which has
multiple complementary regions in the same mRNA
are shown (Table 1: line 5).
• Strand selection: This filter extracts
complementarities between the gene and either the
sense or the antisense strand of the siRNA.
• Specific gene or a list of genes: The list of off-
target candidates can be filtered for one or more
genes of interest. Such genes could be for example
the known top hits of the screen.
Figure 3: General schema the workflow representing
Filtering possibilities for off-target candidates through
different options which correspond to type of off-targets in
Table 1 ines 1, 2, 4, 5, 6, 7.
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
256

Based on this objective, a software toolkit to
analyse RNAi screening data for off-target effects
has been implemented. This toolkit was integrated
into the High Content Data Chain (HCDC)
environment, which is an extension of the Konstanz
Information Miner (KNIME) platform. The main
aim of the implementation was to develop nodes as
flexible and user-friendly as possible. Consequently,
the software application to analyse off-target effects
implemented in HCDC is presented as a set of
nodes.
2.3 Workflows - Execution of
Individual Analyses
The methods and tools described above in
combination with the existing nodes provided from
HCDC and KNIME represent the toolkit for creating
individual workflows. To demonstrate how an
analysis of off-target effects can be performed, an
exemplary workflow is shown in Fig. 4. Node
settings used for this workflow are described on
webpage (HCDC).
Figure 4: Filter combinations in a workflow to analyze
off-target effects.
The filtering steps are of great importance in the
analytic process, because they help to remove false
positives from the output table and to find the most
probable off-target genes. As mentioned before, the
filter nodes can be used and combined completely
flexibly. This is demonstrated in Figure 4 showing
three sample workflows for performing meaningful
filtering.
It has been reported that siRNAs can cause off-
target effects because of their miRNA-like
behaviour (Doench et al., 2003). This means that
there is a seed region complementarity with the
3’UTR of the mRNA. The effect could be amplified
if this complementary region occurs multiple times
in the 3’ end of the mRNA. To filter for these off-
target effects, the filter nodes shown in variant A of
Figure 4 can be connected. The Alignment Filter
gets the results which show a stretch of matches at
the seed region (positions 13-18). Afterwards, the
Location Filter reduces the table to those alignments
located in the 3’UTR of the mRNA, and finally the
Multiple Hit Filter shows only the results where a
siRNA has at least 2 complementary regions in an
mRNA. This
example demonstrates the advantage of
the workflow environment. The filtered output tables
are always available for each node enabling the user
to compare the results after each filtering step.
Variant B in Figure 4 shows that also combinations
of the same type of filtering node are allowed. The
first Alignment Filter, for example, only displays the
alignments, which contain a match at the positions
9-11 and 13-18. Additionally, the second Alignment
Filter node reduces this output table to those
alignments having at least 11 matches in total. As a
last example, a very common filter combination to
analyse off-target effects is shown in variant C of
Figure 4. Off-target effects triggered by a
complementary region with a top hit gene of the
screen are normally of great interest. The occurrence
of such off-target effects would explain unexpected
phenomena in screening results. Therefore, the Text
Filter node can be used with a list of top hit gene
symbols so that only siRNA results showing a
complementary region with a top hit are included in
the output table afterwards. However, not all of
these results are correct off-target candidates and
further filtering nodes have to be applied to get the
most probable ones.
3 RESULTS
For the purpose of validating the new toolkit and
showing its usability in actual research projects, it
has been applied to experimental data from High
Content Screenings technology. The first dataset
used for validation is a RNAi screen for components
involved in ribosome biogenesis. Ribosomes are
macromolecular complexes that synthesize proteins.
Ribosomes are composed of a small and a large
subunit, which both consist of ribosomal proteins
and RNA. In eukaryotes, the biogenesis of these
subunits is a complex multistep process including
the assembly of different components into the
subunits in the nucleolus, the export of these
precursors to the cytoplasm, and final cytoplasmic
maturation steps. In this project, the biogenesis of
the small ribosomal subunit (40S subunit) was
studied in human cells by performing a genome-
wide siRNA screen. The Rps2-YFP assay used
(Wild et al., 2010) enables the visual detection of
nuclear 40S maturation defects upon depletion of a
NewAlgorithmforAnalysisofOff-targetEffectsinsiRNAScreens
257

Table 2: First test set applied to the Seed-Motif-Search Node (default parameters), the Alignment filter (matches at position
5-18) and the Threshold Filter (-94). The first 4 columns describe: 1- gene name, 2- GeneID, 3- RefSeq, 4- gene
description. The last 3 columns provide the alignment score of siRNA and mRNA and the position of the alignment on the
mRNA.
protein by RNAi. As a numerical readout, a hit rate
was determined for each siRNA, measuring the
amount of cells displaying ribosome synthesis
defects. In total, 17632 genes and 5318 predicted
genes where each gene is target by four oligos.
In a first step, 13 siRNAs of the first dataset
were selected such that one siRNA has a hit rate >
0.9, while the other three siRNAs targeting this gene
have a hit rate < 0.03. Applying these criteria yields
many genes, which constitute potential off-target
hits. Less stringent filtering criteria may be applied
to get a longer list of potential off-target hits.
However, for the purpose of this software validation
analysis, it seems beneficial to visualize the off-
target analysis of a small set of genes. To find
complementary regions between the siRNAs
(targeting the selected genes) and all human
mRNAs, the Seed-Motif-Search is used. Since the
resulting list has a size of 477968 rows, a large
number of filter combinations is applied afterwards
to reduce its size and to detect the most probable off-
target candidates for an siRNA. Depending on the
combination of different filter nodes the possible
off-target genes vary considerably. An example of a
strict filtering is the use of the Alignment Filter
which allows only alignments containing matches at
position 5-18. The output of this node is shown in
Table 2.
All obtained potential off-target genes seem to be
good off-target candidates since they show a high
percentage of complementarity with the respective
siRNA.
For one gene (DTNBP1) a potential off-target is
RPS3, a ribosomal protein required for 40S
biogenesis and a top hit in the provided dataset.
Hence, an off-target effect of the siRNA
targeting DTNBP1 against RPS3 might explain the
high hit rate observed for this siRNA.
There is no fixed score that would give a clear
suggestion for potential off-target. Nevertheless,
after several observations and analyses we can
suggest that an alignment score above 73 with a
match length minimum of 14 nt will predict a
potential off-target with a reasonable fidelity.
3.1 Validation
To experimentally test the off-target prediction
results (Table 2), we analysed RPS3 levels upon
RNAi against DTNBP1. Using the same readout as
in the screen (nuclear accumulation of Rps2-YFP),
we tested all 4 siRNAs against DTNBP1 present in
the genome-wide screen. Additionally, we measured
RPS3 levels by Western blotting (Figure 5). As
observed in the genome-wide screen, si-DTNBP-2
treatment leads to 40S biogenesis defects
comparable to si-RPS3 treatment. In contrast, the
other siRNAs against DTNBP1 cause no 40S
biogenesis defect (comparable to untreated and si-
control sample). Importantly, RPS3 levels were
significantly reduced upon si-DTNBP1-2 treatment,
but unaffected by the other siRNAs against
DTNBP1. Therefore, the high hit rate of si-
DTNBP1-2 is likely caused by RPS3 depletion and
hence, as predicted, an off-target effect.
Figure 5: Experimental verification of predicted off-target
effect.
A HeLa Rps2-YFP cells were treated with
indicated siRNAs according to Wild et al. Images
were taken with epi-fluorescence microscopy.
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
258

B Protein levels of RPS3 and ENP1 (as a loading
control) derived from experiment shown in A were
detected by Western blotting. Note that si-DTNBP1-
2 reduces RPS3 levels.
4 CONCLUSIONS
The major objective of this paper was to describe a
new algorithm and software toolkit to analyse off-
target effects in RNAi screening data.
The test and validation phase has proven that the
software already provides a powerful and flexible
toolkit for analysing off-target effects. Testing and
validating the toolkit with an actual high content
dataset revealed that the workflow environment is
suitable for off-target analysis. The analyses of the
given dataset show that the environment allows for a
dynamic workflow adaption based on intermediate
results, e.g. by supplemental Text Filter integration.
In addition to the flexible workflow creation facility,
the individual configuration options of a single node
are also advantageous. All in all, the software
environment with its flexibility turns out to be very
suitable to analyse off-target effects in RNAi
screening data. An important aspect is the reliability
of the results obtained in the analysis process. In this
case the results seem to be reasonable and correct.
We would like emphasize that our predictions
neither include the effects of siRNA concentration
nor do they attempt to account for the siRNA pool
constructs. It is clear that both these effects are of
critical practical consequence and that a
computational model supporting them is desirable.
At the moment, however, there is insufficient
published data on the efficacies of pools to be able
to construct a high-confidence model of pool effects.
REFERENCES
Alemán, L. M., Doench, J. G. and Sharp, P. A., 2007.
Comparison of sirna-induced off-target rna and protein
effects. RNA, 13(3):385–95.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W and
Lipman, D. J., 1990. Basic local alignment search tool.
J Mol Biol, 215(3):403–10.
Amarzguioui, M., Holen,T., Babaie, E. and Prydz, H.
2003. Tolerance for mutations and chemical
modifications in a sirna. Nucleic Acids Res,
31(2):589–95.
Anderson, E. M., Birmingham, A., Baskerville, S.,
Reynolds, A., Maksimova, E., Leake, D., Fedorov, Y.,
Karpilow, J. and Khvorova, A., 2008. Experimental
validation of the importance of seed complement
frequency to sirna specificity. RNA, 14(5):853–61.
Birmingham, A., Anderson, E. M., Reynolds, A., Ilsley-
Tyree, D., Leake, D., Fedorov, Y., Baskerville, S.,
Maksimova, E., Robinson, K., Karpilow, J., Marshall,
W.S and Khvorova, A., 2006. 3’ utr seed matches, but
not overall identity, are associated with rnai off-
targets. Nat Methods, 3(3):199–204.
Doench, J. G., Petersen, C. P. and Sharp, P. A., 2003.
Sirnas can function as mirnas. Genes Dev, 17(4):438–
42.
Elbashir, S. M, Martinez, J., Patkaniowska, A., Lendeckel,
W. and Tuschl, T., 2001. Functional anatomy of sirnas
for mediating efficient rnai in drosophila melanogaster
embryo lysate. EMBO J, 20(23):6877–88.
Fedorov, Y., King, A., Anderson, E. M., Karpilow, J.,
Ilsley, D., Marshall, W. S. and Khvorova, A., 2005.
Different delivery methods-different expression
profiles. Nat Methods, 2(4):241.
Fedorov, Y., Anderson, E. M, Birmingham, A., Reynolds,
A., Karpilow, J., Robinson, K., Leake, D., Marshall,
W.S. and Khvorova, A., 2006. Off-target effects by
sirna can induce toxic phenotype. RNA, 12(7):1188–
96.
Grillo, G., Turi, A., Licciulli, F., Mignone, F., Liuni, S.,
Banfi, S., Gennarino, V. A., Horner, D. S., Pavesi, G.,
Picardi, E., Pesole, G., 2010. UTRdb and UTRsite
(RELEASE 2010): a collection of sequences and
regulatory motifs of the untranslated regions of
eukaryotic mRNAs. Nucleic Acids Res., 38, 75-80.
Hannon, H. J., 2002. Rna interference. Nature,
418(6894):244–51, July.
Haley, B. and Zamore, P. D., 2004. Kinetic analysis of the
rnai enzyme complex. Nat Struct Mol Biol, 11(7):599–
606.
HCDC web page, bioinformatics module:
[http://hcdc.ethz.ch/index.php?view=article&id=25]
Jackson, A. L, Burchard, J., Leake,D., Reynolds, A.,
Schelter, J., Guo, J., Johnson, J. M., Lim, L.,
Karpilow, J., Nichols, K., Marshall, W. S., Khvorova,
A. and Linsley, P. S., 2006. Position-specific chemical
modification of sirnas reduces "off-target" transcript
silencing. RNA, 12(7):1197–205.
Jackson, A. L., Burchard, J., Schelter, J., Chau, B. N.,
Cleary, M., Lim, L. and Linsley, P. S., 2006.
Widespread sirna "off-target" transcript silencing
mediated by seed region sequence complementarity.
RNA, 12(7):1179–87.
Jackson, A. L., Bartz, S. R., Schelter, J., Kobayashi, S. V.,
Burchard, J., Mao, M., Li, B., Cavet, G. and Linsley,
P. S., 2003. Expression profiling reveals off-target
gene regulation by rnai. Nat Biotechnol, 21(6):635–7.
Lewis, B. P., Burge, C. B. and Bartel, D. P., 2005.
Conserved seed pairing, often flanked by adenosines,
indicates that thousands of human genes are microrna
targets. Cell, 120(1):15–20.
Lin, X., Ruan, X., Anderson, M. G., McDowell, J. A.,
Kroeger, P. E., Fesik, S. W. and Shen, Y., 2005. Sirna-
mediated off-target gene silencing triggered by a 7 nt
complementation. Nucleic Acids Res, 33(14):4527–35.
NewAlgorithmforAnalysisofOff-targetEffectsinsiRNAScreens
259

Saxena, S., Jónsson, Z.O. and Dutta, A. 2003. Small rnas
with imperfect match to endogenous mrna repress
translation. implications for off-target activity of small
inhibitory rna in mammalian cells. J Biol Chem,
278(45):44312–9.
Semizarov, D., Frost, L., Sarthy, A., Kroeger, P.A,
Halbert, D. N. and Fesik, S. W., 2005. Specificity of
short interfering rna determined through gene
expression signatures. Proc Natl Acad Sci U S A,
100(11):6347–52.
Smith, T. F. and Waterman, M. S., 1981. Identification of
common molecular subsequences. J Mol Biol,
147(1):195–7.
Ui-Tei, K., Naito, Y., Nishi, K., Juni, A. and Saigo, K.,
2008. Thermodynamic stability and watson-crick base
pairing in the seed duplex are major determinants of
the efficiency of the sirna-based off-target effect.
Nucleic Acids Res, 36(22):7100–9.
Wild, T., Horvath, P., Wyler, E., Widmann, B.,
Badertscher, L., Zemp, I., Kozak, K., Csusc, G., Lund,
E., Kutay, U., (2010) A protein inventory of human
ribosome biogenesis reveals an essential function of
Exportin 5 in 60S subunit export. PLoS Biol,
8:e1000522.
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
260
