
Hadoop-RINS
A Hadoop Accelerated Pipeline for Rapid Nonhuman Sequence Identification
Li Jiangyu
1,2
, Liu Yang
1
, Wang Xiaolei
1
, Mao Yiqing
1
, Wang Yumin
2
and Zhao Dongsheng
1
1
Institute of Health Service and Medical Information, Academy of Military Medical Sciences, Taiping Road, Beijing, China
2
Institute of Biotechnology, Academy of Military Medical Sciences, East Avenue, Beijing, China
Keywords: High-Throughput Sequencing, Metagenomics, RINS, Hadoop, MapReduce.
Abstract: Sequencing data increase rapidly in recent years with the development of high-throughput sequencing
technology. Using parallel computing to accelerate the computation is an important way to process the large
volume of sequence data. RINS is a pipeline used to identify nonhuman sequences in deep sequencing
datasets. It uses user-provided microbial reference genomes to reduce the number of reads to be processed
and improve the processing speed. But all of its steps run serially. As a result, the processing speed of RINS
slows down sharply as the sequencing data and reference genomes increase. In this article, we report a
pipeline that processes sequencing data parallel through Hadoop. By comparing the runtime using same
dataset, Hadoop-RINS is proved to be significantly faster than RINS with the same computation result.
1 INTRODUCTION
With the development of high throughput
sequencing technology, researches that focus on
sequencing data processing keep on increasing. As a
result, researches based on high-throughput
sequencing technology have advanced greatly in
recent years, such as Metagenomics and RNA-Seq.
Many human diseases are caused by pathogens,
which are difficult to detect with traditional
methods. High-throughput sequencing presents a
possible way to identify these pathogens (Kostic et
al., 2011). So researches on applications of pathogen
identification with high-throughput sequencing have
become a hot spot in sequencing data processing. At
present, a main topic in the application research is to
design pipelines for high-throughput sequencing
data processing. The main function of the pipelines
is to filter unrelated sequences and analyze
remaining reads.
There are two methods in the design of these
pipelines. The first method filters the sequencing
data by aligning them to different reference genomes
and maps the remaining reads to find the pathogens
in the sample. The second method assumes the
species of pathogens in the sample with prior
knowledge and processes the sequencing data by
aligning them to the genomes of assumed species,
and then uses the result to verify the assumption.
PathSeq (Kostic et al., 2011) is an example of the
first method. It aligns the sequencing data to human
genome and filters reads that are similar, and then
classifies the nonhuman reads by aligning them to
microbial genome database. PathSeq is very suitable
to discover unknown microbe. The second method is
used in RINS (Bhaduri et al., 2012). RINS aligns the
sequencing data to assumed reference genomes
which are based on prior knowledge, and filters out
the reads with similarity lower than given threshold,
then maps the remaining reads to human genome
and collects the reads unmapped for assembly,
finally extends the contigs using original sequencing
data and aligns contigs to the assumed reference
genomes to verify the assumption. RINS is suitable
to identify samples with prior knowledge of species.
Utilizing prior knowledge, we can reduce the
data scale and further reduce data processing time.
In cases when prior knowledge exists, RINS is
generally more effective than PathSeq. But RINS
follows an assume-verify diagram which means it
has to assume several times before the assumption
proves to be right. What’s more, the runtime of
RINS increases quickly with the increasing volume
of sequencing data. The pipeline of RINS is serial.
We can improve the processing speed of RINS by
parallelizing the pipeline. MapReduce is a suitable
model for our parallelization work.
296
Jiangyu L., Yang L., Xiaolei W., Yiqing M., Yumin W. and Dongsheng Z..
Hadoop-RINS - A Hadoop Accelerated Pipeline for Rapid Nonhuman Sequence Identification.
DOI: 10.5220/0004239602960299
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2013), pages 296-299
ISBN: 978-989-8565-35-8
Copyright
c
2013 SCITEPRESS (Science and Technology Publications, Lda.)

2 MAPREDUCE MODEL
MapReduce is the most popular programming model
for processing large data sets (Lei, 2011). It is
typically used to do distributed computing on
clusters of computers (Stephen, 2008). In the
MapReduce model, programs are divided into map
phase and reduce phase. During the map phase, data
are partitioned into several blocks and are processed
separately. In the reduce phase, data are collected.
There are several implementations of MapReduce
model. A popular implementation is Apache
Hadoop. Hadoop is an open-source software
framework that supports data-intensive distributed
applications. It can process large scale data
distributedly. As a result, it is the preferred choice
for many cloud computing applications.
Hadoop is widely used in high-throughput
sequencing data processing. It is used in PathSeq to
distribute sequencing data and process them in
parallel. Hadoop-BLAST (Nachankar and Arvind,
2011) uses Hadoop to efficiently distribute tasks and
reliably transmit data in sequence alignment process.
Cloud-MAQ (Talukder et al., 2010) makes MAQ
parallel and scalable through Hadoop and enhances
the performance of MAQ significantly.
3 METHODS
3.1 Workflow of RINS and Bottleneck
Analysis
The workflow of RINS is shown in Figure 1. The
first step is to check the sequencing data format and
divide the reads into k-mers. For FASTQ data, we
need to transform it to FASTA format first. The
second step is to map the k-mers to reference
genomes by running BLAT (Kent, 2002) and extract
reads with similarity scores higher than given
threshold. The third step is to compress the reads by
filtering the duplicate reads. The fourth step is to run
Bowtie to align reads to human genome and extract
unmapped reads. The fifth step is to assemble
contigs with Trinity (Grabherr et al., 2011). The last
step runs BLAST (Altschul et al., 1990) to map
nonhuman contigs to reference genomes and verify
the assumption. In the workflow only step 5 can't
run in parallel. We first ran RINS on our platform
and analysed the bottleneck using the runtime of
each step.
Server: eight servers running CentOS 6.2, each
with a quad-core 2.299 GHz AMD CPU and 4 GB
RAM.
Figure 1: Workflow of RINS.
Datasets: Human genome version hg19 which is
downloaded from UCSC. Virus genomes that were
downloaded from NCBI in Oct 2011 are used as
reference genomes. We use SRR073726 and
SRR073732 as the test data. SRR073726 is also used
in the original literature of RINS as test data. The
two datasets are paired-end reads and can be
downloaded from NCBI.
To eliminate the possible impact of differences
among the eight servers, we randomly selected four
servers to process the two datasets by RINS and
performed four tests. The runtime of each step is
listed in Table1 and Table 2.
Table 1: Runtime of each step in RINS (SRR073726).
Step
Runtime (sec)
test 1 test 2 test 3 test 4 avg.
step 1 1177 1169 1256 1228 1208
step 2 4602 4541 4634 4682 4615
step 3 204 134 164 179 170
step 4 179 158 193 225 189
step 5 569 648 620 585 606
step 6 4 5 4 4 4
total 6745 6655 6871 6903 6794
Table 2: Runtime of each step in RINS (SRR073732).
Step
Runtime (sec)
test 1 test 2 test 3 test 4 avg.
step 1 1140 1188 1244 1272 1211
step 2 6608 6707 6803 6826 6736
step 3 119 139 125 132 129
step 4 103 121 111 120 114
step 5 598 604 602 593 599
step 6 4 4 4 5 4
total 8572 8762 8889 8948 8793
In above result the most time-consuming steps of
RINS are step 2, step 1 and step 5, which account for
nearly 95% of the total runtime. These three steps
are the bottleneck of RINS. By reducing the runtime
of them we can improve the processing speed.
Hadoop-RINS-AHadoopAcceleratedPipelineforRapidNonhumanSequenceIdentification
297
3.2 Parallelization of RINS
After analysing the steps in RINS, we find that in the
first step data file is read from top to bottom in one
pass during the process of format transformation,
there is no need to parallelize the process. In step 5,
the original sequencing data and the entire filtered
nonhuman reads are needed to assemble contigs, it's
difficult to parallelize the process, but alignment in
this step can run in parallel. Inputs of the other steps
have low data coupling, so these steps are suitable
for parallelization. We use Hadoop to parallelize
these steps. The paralleled pipeline is called
Hadoop-RINS for short.
3.3 Pipeline of Hadoop-RINS
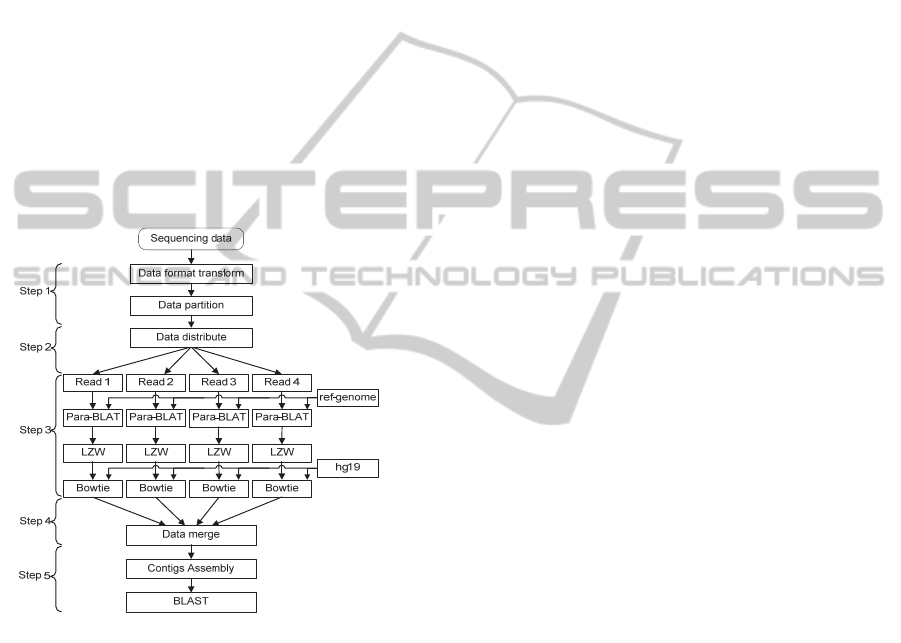
Pipeline of Hadoop-RINS is depicted in Figure 2. It
is implemented through the following steps.
Figure 2: Pipeline of Hadoop-RINS.
1) Data Preprocessing: We write the data format
transformation and partition program with Perl. The
program runs on the master node of the Hadoop
cluster. We need to check the format of sequencing
data at first. The FASTA file is partitioned directly,
and the FASTQ file is partitioned during the data
format transformation. For paired-end data, the
mated two files should be cut at the same line to
avoid mismatch between the paired reads.
2) Data Distribution: In this step, all the partitioned
sequencing data files are distributed to the Hadoop
slave nodes. The number of task nodes is equal to
the partition number of sequencing data.
3) Map Phase: We use a script to execute the work
of map phase serially. The script runs on every slave
node of Hadoop. After the assigned data file is
copied to the node, the script divides the sequencing
data into k-mers. Then BLAT is used to map each k-
mers to the reference genomes. As BLAT is time-
consuming, we developed a parallel BLAT to make
the most of CPU. Then, we extract reads with higher
relevance score than the given threshold and use the
LZW(
Welch, 1984) algorithm to filter the duplicate
reads in the extracted data. At last, we align these
filtered reads to hg19 by Bowtie and reserve reads
that are not mapped.
4) Reduce Phase: We collect the results generated
in map phase to Hadoop master node, and merge
these nonhuman reads into a single file.
5) Final Result: A script is written to finish the
work in step 5 and step 6 of RINS. The merged data
file is assembled by Trinity, and then the contig file
is copied to the computing nodes. All the sequencing
data partitions distributed in data distribution are
aligned to these contigs by BLAT. We use the
alignment result to extend the contigs. Finally, we
merge all the alignment results from the computing
nodes into one file and run BLAST with nonhuman
reference genomes to find the origin of these contigs.
4 RESULTS
In order to evaluate the performance of Hadoop-
RINS, we designed four groups of experiments to
measure the runtime of Hadoop-RINS and RINS.
SRR073726 and SRR073732 were test data. The
first group ran RINS on single node. The second
group ran Hadoop-RINS on two nodes. The two
nodes were selected randomly from the cluster, and
one node was configured as master node and slave
node, the other was slave node. The third group ran
Hadoop-RINS on four nodes. We randomly select
four nodes in each test, and one node acted as master
node and slave node, the other three nodes were
slave nodes. The forth group ran Hadoop-RINS on
all eight nodes. One node was configured as master
node and slave node, and the other nodes were slave
node. For each dataset, test repeated four times.
Table 3 and Table 4 present the average runtime
for the two datasets respectively. Hadoop-RINS
achieves significant speedup compared with RINS.
For SRR073726 (SRR073732), Hadoop-RINS
running on 2-node cluster is about 3.58x (3.54x)
times faster than RINS, while processing speed on 4-
node cluster is about 4.88x (5.36x) times faster than
RINS and processing speed on 8-node cluster is
about 5.75x (6.34x) times faster than RINS.
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
298

Table 3: Runtime of four tests (SRR073726).
Test
Number
Runtime (sec)
RINS on
single node
2-node
cluster
4-node
cluster
8-node
cluster
test 1 6745 1889 1338 1152
test 2 6655 2059 1488 1222
test 3 6871 1856 1437 1134
test 4 6903 1797 1308 1215
avg. 6794 1900 1393 1181
Table 4: Runtime of four tests (SRR073732).
Test
Number
Runtime (sec)
RINS on
single node
2-node
cluster
4-node
cluster
8-node
cluster
test 1 8572 2571 1624 1392
test 2 8762 2563 1722 1429
test 3 8889 2409 1638 1317
test 4 8948 2381 1584 1405
avg. 8793 2481 1642 1386
Table 5 shows the runtime of each step in
Hadoop-RINS. The runtime of step 3 decreases
significantly as the number of computing nodes
increases. Due to partly paralleled, the runtime of
step 5 also decreases as the number of nodes
increases. While the runtime of file format
transformation and file segmentation changes little.
The runtime in file distribution increases with the
number of computing nodes. The runtime of step 4
seems inconsistent with the increase of nodes.
Table 5: Average runtime of each step in Hadoop-RINS
(SRR073726/SRR073732).
Step
Runtime (sec)
2 nodes 4 nodes 8 nodes
step 1 224/327 222/345 307/354
step 2 59/80 87/151 116/137
step 3 1368/1691 753/932 441/510
step 4 3/108 180/42 178/264
step 5 247/274 151/171 139/121
total 1901/2479 1393/1641 1181/1386
BLAT is the main bottleneck in RINS. We have
tried to run BLAT with divided data and the runtime
of Hadoop-RINS reduces greatly. From the runtime
of multi nodes cluster, we can see the speedup does
not increase remarkably with node number. That's
because the runtime of step 1 and step 2 does not
decrease with the increase of nodes. So the runtime
proportion increases with the number of nodes. For
the 8-node cluster, it can amount to 35.8%.
The inconsistence of step 4 is caused by the
differences of computing nodes. For some reason the
processing speed of one node is slower than others,
then Hadoop needs to wait until all nodes finish their
work, which increases the runtime of step 4. So in a
heterogeneous cluster, the runtime may be
influenced by the slowest node. Heterogeneous
environment is not recommended for Hadoop-RINS.
Compared with RINS, Hadoop-RINS running on
2-node cluster, 4-node cluster and 8-node cluster get
the same contigs as those of RINS. So Hadoop-
RINS has the same accuracy with RINS.
5 DISCUSSION
Processing speed is an important indicator in
pathogen detection. In this article, we analyze the
pipeline and runtime of RINS to find the bottleneck,
and then we use Hadoop to realize a parallel pipeline
to finish the main steps of RINS. In the future, we
will implement a sub-pipeline to analyze the filtered
data which can’t be mapped to reference genomes.
REFERENCES
Altschul, S. F. et al., 1990. Basic local alignment search
tool. J Molecular Biology. 215(3):403-410.
Bhaduri, A. et al., 2012. Rapid Identification of
Nonhuman Sequences in High Throughput Sequenc-
ing Data Set. Bioinformatics. 28(8):1174-1175.
Grabherr, M. G. et al., 2011. Full-length transcriptome
assembly from RNA-seq data without a reference
genome. Nature Biotechnology. 29(7):644-652.
Kent, W. J., 2002. BLAT--the BLAST-like alignment tool.
Genome research. 12(4): 656-664.
Kostic, A. D. et al., 2011. PathSeq: software to identify or
discover microbes by deep sequencing of human
tissue. Nature Biotechnology. 29(5):393-396.
Lei W. Y., 2011. Cloud-computing: the strategy and
practice of enterprise information construction.
http://blog.sina.com.cn/s/blog_6c5cffe30100p833.html.
Nachankar, V., Arvind, D., 2011. Hadoop-BLAST.
http://salsahpc.indiana.edu/csci-b649-2011/collection/
project1/report/group11_proj1_report.pdf.
Stephen S., 2008. Google spotlights data center inner
workings. http://news.cnet.com/8301-10784_3-99551
84-7.html.
Talukder, A. K. et al., 2010. Cloud-MAQ: the cloud-
enabled scalable whole genome reference Assembly
application. WOCN 2010.
Welch, T. A., 1984. A technique for high-performance
data compression. Computer. 17: 8-19.
Hadoop-RINS-AHadoopAcceleratedPipelineforRapidNonhumanSequenceIdentification
299
