
The Disulfide Connectivity Prediction with Support Vector Machine and
Behavior Knowledge Space
Hong-Yu Chen
1
, Chang-Biau Yang
1,†
, Kuo-Tsung Tseng
2,‡
and Chiou-Yi Hor
1
1
Department of Computer Science and Engineering, National Sun Yat-sen University, Kaohsiung 80424, Taiwan
2
Department of Information Management, Fooyin University, Kaohsiung 83102, Taiwan
Keywords:
Disulfide Bond, Cysteine, Connectivity Pattern, Support Vector Machine, Behavior Knowledge Space.
Abstract:
A disulfide bond, formed by two oxidized cysteines, plays an important role in the protein folding and structure
stability, and it may regulate protein functions. The disulfide connectivity prediction problem is to reveal the
correct information of disulfide connectivity in the target protein. It is difficult because the number of possible
patterns grows rapidly with respect to the number of cysteines. In this paper, we discover some rules to
discriminate the patterns with high accuracy in various methods. Then, we propose the pattern-wise and pair-
wise BKS (behavior knowledge space) methods to fuse multiple classifiers constructed by the SVM (support
vector machine) methods. Furthermore, we combine the CSP (cysteine separation profile) method to form our
hybrid method. The prediction accuracy of our hybrid method in SP39 dataset with 4-fold cross-validation is
increased to 69.1%, which is better than the best previous result 65.9%.
1 INTRODUCTION
A disulfide bond, also called SS-bond or SS-bridge,
is a single covalent bond, and it is usually formed
from the oxidation of two thiol groups. In proteins,
only the thiol groups of cysteine residues can form the
disulfide bonds by oxidation. The goal of the disulfide
connectivity prediction (DCP) problem is to figure out
which cysteine pair would be cross-link from all pos-
sible candidates. It may be conducive to the solution
of the protein structure prediction problem if precise
disulfide connectivity information is available.
There are two main ways for solving the DCP
problem in previous works, pair-wise and pattern-
wise. The pair-wise method focuses on the bonding
potential of each cysteine pair, and encodes the target
according to cysteine pairs. The pattern-wise method
makes a comprehensive survey of the whole connec-
tivity pattern and it usually ranks the connectivity pat-
terns, so the prediction ability may be limited to the
diversity of patterns in a training set.
The pattern-wise DCP task is difficult because
the number of possible connectivity patterns grows
rapidly with respect to the number of cysteines. The
number of possible patterns is given as follows:
N =
C
2B
2
×C
2B−2
2
× . . . ×C
2
2
B!
= (2B− 1)!! (1)
where B is the number of disulfide bonds in the pro-
tein. For instance, if the oxidized state of each cys-
teine is known in advance, then N = 945 when B = 5,
and N is up to 10395 when B = 6. Thus, most studies
restrict the number of disulfide bonds to be from two
to five.
Some statistical analysis (Paul M. Harrison and
Michael J. E. Sternberg, 1994; Chih-Hao Lu et al.,
2007; Leonid A. Mirny and Eugene I. Shakhnovich,
1996; Rotem Rubinstein and Andras Fiser, 2008)
have been applied to the DCP problem. Many re-
searchers tried to solve the problem with machine
learning methods such as neural network (NN) (Pierre
Baldi et al., 2005; Jianlin Cheng et al., 2006; Piero
Fariselli et al., 1999; F. Ferre and P. Clote, 2005;
Pier Luigi Martelli et al., 2002; Alessandro Vullo
and Paolo Frasconi, 2004; Castrense Savojardo et al.,
2013) and support vector machine (SVM) (Yu-Ching
Chen et al., 2004; Yu-Ching Chen and Jenn-Kang
Hwang, 2005; P. Frasconi et al., 2002; Jayavardhana
Rama G. L. et al., 2005; Hsuan-Liang Liu and Shih-
Chieh Chen, 2007; Chih-Hao Lu et al., 2007; Chi-
Hung Tsai et al., 2005; Marc Vincent et al., 2008).
Before 2005, many studies (Pierre Baldi et al.,
2005; F. Ferre and P. Clote, 2005) were devoted to
the DCP problem, but most of their accuracies are
less than 50%. In 2005, Zhao et al. (East Zhao
et al., 2005) utilized the global information in a pro-
112
Chen H., Yang C., Tseng K. and Hor C..
The Disulfide Connectivity Prediction with Support Vector Machine and Behavior Knowledge Space.
DOI: 10.5220/0004541501120118
In Proceedings of the International Conference on Knowledge Discovery and Information Retrieval and the International Conference on Knowledge
Management and Information Sharing (KDIR-2013), pages 112-118
ISBN: 978-989-8565-75-4
Copyright
c
2013 SCITEPRESS (Science and Technology Publications, Lda.)

tein, called cysteine separation profile (CSP), which
is the separations among all oxidized cysteines on a
protein sequence.
In the past, the bonding states of each cysteine
pair are usually used to describe the disulfide pattern
and used as the samples of SVM. Lu et al. (Chih-
Hao Lu et al., 2007) call this type of representation
of the disulfide pattern as the CP
1
representation. In
2007, Lu et al. further proposed a novel concept of
the CP
2
representation which use every two cysteine
pairs (four cysteines) as the samples, and applied the
genetic algorithm (GA) to the optimization of feature
selection.
In 2012, Wang et al. (Chong-Jie Wang et al.,
2012) proposed a hybrid model based on SVM and
the weighted graph matching (Piero Fariselli and Rita
Casadio, 2001), with accuracy 65.9%. They extracted
different feature sets depending on whether the num-
ber of disulfide bonds in a protein is odd or even. The
main difference of feature sets for the two submod-
els is the secondary structure information around the
oxidized cysteines.
The rest of this paper is organized as follows. We
introduce some preliminary knowledge, including re-
lated tools and previous works of the DCP problem
in Section 2. In Section 3, we describe our hybrid
method for solving the DCP problem. Our experi-
mental results are shown in Section 4, and we also
compare the prediction accuracy of our method with
the previous works. Finally, our conclusion are given
in Section 5.
2 PRELIMINARY
In this section, we introduce some background knowl-
edge for this paper, including the Position-Specific
Scoring Matrix (PSSM), support vector machine
(SVM) and behavior knowledge space (BKS).
2.1 Position-specific Scoring Matrix
Position-Specific Scoring Matrix (PSSM) (Stephen F.
Altschul et al., 1997), also called profile, is a scor-
ing matrix derived from a group of aligned protein se-
quences. It represents the similarity of residues in ev-
ery specific position of a query sequence (a target pro-
tein) according to the alignment result of the query se-
quence and the others (probes) in database. Basically,
PSSM is a matrix of size N × 20, where N denotes
the length of a query sequence and every residue in
the query sequence contains a 20-element vector. The
20-element vector respectively represents the scores
of 20 standard amino acids which are substituted for
the position-specific residue of the query sequence.
2.2 Support Vector Machine
Support vector machine (SVM) is a machine learning
method for classification and regression. It was first
introduced by Vapnik (Vladimir N. Vapnik, 1999) in
1999. SVM seeks to create a hyperplane to discrimi-
nate different labels of the data elements (vectors) in
the training set and utilizes the model to predict the la-
bels of target data elements. Each vector is considered
as a point in the feature space, and each dimension of
the coordinates represents one kind of features. To
discover the discriminative features is the key point
of SVM.
For SVM implementation in this paper, we use the
LIBSVM
package (Chih-Chung Chang and Chih-Jen
Lin, 2001), which is an easy-to-use tool for support
vector classification (SVC) and support vector regres-
sion (SVR). The SVC function classifies the data with
their probabilities, and the SVR function generates
the regression value of each target data element.
2.3 Behavior Knowledge Space
Behavior knowledge space (BKS) (Sarunas Raudys
and Fabio Roli, 2003) is a method for fusing multi-
ple classifiers. It builds a look-up table for estimat-
ing the posterior probabilities and every combination
of votes. Assume there are m classifiers composing
an ensemble for a classification task of n labels. The
BKS table contains n
m
entries, the number of all pos-
sible combinations of m classifiers’ outputs. And each
entry records the distribution of n true labels in the
training set.
Table 1 illustrates an example of the BKS table for
a 3-label classification problem with two classifiers.
The ’C1’ and ’C2’ represent the outputs from the two
classifiers, and the entries below them show all nine
possible prediction combinations. Cells below ’Real
label’, ’L1’, ’L2’, and ’L3’, are the distribution of the
true labels associated with the predicted label vec-
tors. For example, when ’C1’=’L1’ and ’C2’=’L3’,
the predicted answer of the ensemble is ’L2’ since it
is the most possible label. As another example, if we
have ’C1’=’L3’ and ’C2’=’L2’, the answer goes to
’L3’.
3 ALGORITHMS FOR
CONNECTIVITY PREDICTION
The prediction accuracies of Chung et al. (Wei-Chun
TheDisulfideConnectivityPredictionwithSupportVectorMachineandBehaviorKnowledgeSpace
113
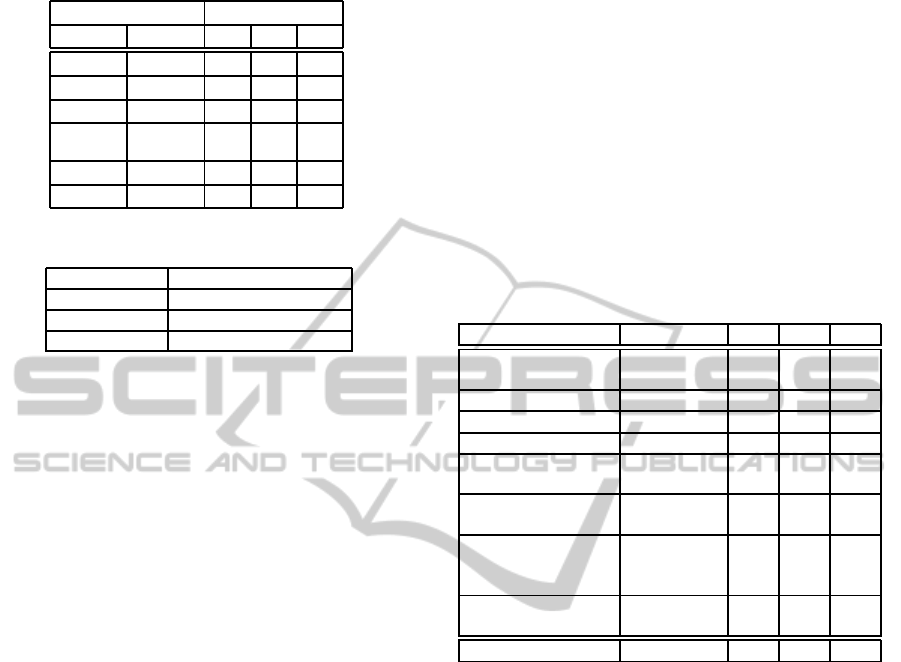
Table 1: An example of the BKS table.
Predicted label Real label
C1 C2 L1 L2 L3
L1 L1 23 8 2
L1 L2 5 0 4
L1 L3 2 7 1
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
L3 L2 1 1 5
L3 L3 1 3 12
Table 2: The feature vector of the permutation order.
Permutations Feature vector
C
1
-C
2
-C
3
-C
4
(0.25, 0.5, 0.75, 1)
C
1
-C
3
-C
2
-C
4
(0.25, 0.75, 0.5, 1)
C
1
-C
4
-C
2
-C
3
(0.25, 1, 0.5, 0.75)
Chung et al., 2009) and Wang et al. (Chong-Jie Wang
et al., 2012) are 63.5% and 65.9%, respectively. It
may be hard to find more features with good discrim-
ination capability for a single SVM method in the
connectivity prediction. However, we may get better
accuracies if we fuse the advantages of the multiple
models.
Our method is based on SVM models, and we use
BKS to fuse these models. The features and cysteine-
pair representation we adopt are inspired by Wang et
al. (Chong-Jie Wang et al., 2012) and Lu et al. (Chih-
Hao Lu et al., 2007). In addition, we also combine
the CSP method (East Zhao et al., 2005) to our hybrid
method.
3.1 Feature Extraction
We follow the features used by Wang et al. (Chong-
Jie Wang et al., 2012) and add the new feature permu-
tation order for the model of CP
2
representation. The
definition of permutation order is given as follows.
Permutation order: This feature implies the order
of feature extraction in each cysteine window. For ev-
ery cysteine-pair combination in the CP
2
representa-
tion, we encode the samples in three permutations il-
lustrated in Table 2. For example,C
1
-C
3
-C
2
-C
4
means
that the first and third cysteines form a disulfide bond
in these four cysteines, and the second and fourth
form the other bond. This bond pattern is represented
by (0.25, 0.75, 0.5, 1).
3.2 SVM Method
We implement three SVM models with different fea-
tures, CP
1
F
521
, CP
1
F
623
and CP
2
Label
2
. Table 3
shows the feature set used in each model. These fea-
tures are encoded by the segments of every cysteine
pair. The cysteine segment is a window centering at
a target cysteine. Many previous works (Yu-Ching
Chen and Jenn-Kang Hwang, 2005; Guantao Chen
et al., 2006; Chao-Chun Chuang et al., 2003; F. Ferre
and P. Clote, 2005; David T Jones, 1999; Jayavard-
hana Rama G. L. et al., 2005; Chih-Hao Lu et al.,
2007; Pier Luigi Martelli et al., 2002; Rotem Rubin-
stein and Andras Fiser, 2008; Chi-Hung Tsai et al.,
2005; Marc Vincent et al., 2008) also adopted the sim-
ilar idea of the window approach. Here we set the
window size to 13.
Table 3: The feature sets used in our three SVM models.
Here, 2k + 1 denotes the window size centering at a target
cysteine, whose value is set to 13 in this paper.
Feature size
M
x
M
y
M
z
Distance of
cysteines
1
Y Y Y
Cysteine order 2
Y
Protein weight 1
Y
Protein length 1
Y
Amino acid
composition
20
Y
PSSM around
cysteine
(2k+1)
×20× 2
Y Y Y
Secondary
structure
around cysteine
(2k+1)
×3× 2
Y
Permutation
order
4
Y
Total size
521 623 525
x
CP
1
F
521
model.
y
CP
1
F
623
model.
z
CP
2
Label
2
model.
3.3 BKS Methods
We adopt the BKS concept to fuse the classifiers men-
tioned above. We design two BKS models, pattern-
wise BKS and pair-wise BKS, combined with the
probability intervals. The probability intervals for
predicting different proteins are illustrated in Table 4.
The pattern-wise BKS is constructed from the
combinations of the predicted pattern probabilities of
two classifiers, CP
1
F
521
and CP
1
F
623
. The pattern-
wise BKS method is used for the prediction of pro-
teins with 2 or 3 bonds. Table 5 illustrates an exam-
ple of the partial pattern-wise BKS table for 2-bond
proteins. For example, in the second row, the proba-
bilities of the predicted pattern 1-1-2-2 for both clas-
sifiers locate in (0.15, 0.2). In this case, 5, 3 and 1
proteins have the true patterns 1-1-2-2, 1-2-1-2 and 1-
2-2-1, respectively. Thus, the predicted answer would
be 1-1-2-2. We set the threshold of the pattern sup-
port in the pattern-wise BKS table to 2, and reject to
KDIR2013-InternationalConferenceonKnowledgeDiscoveryandInformationRetrieval
114

Table 4: The probability intervals for BKS methods with
various numbers of bonds, denoted by B.
B Type of BKS Probability intervals
2 Pattern-wise
(0, 0.15, 0.2, 0.25,
0.3, 0.35, 0.5, 1)
3 Pattern-wise
(0, 0.25, 0.5, 1)
4 Pair-wise
(0, 0.1, 0.2, 0.3,
0.4, 0.5, 1)
5 Pair-wise
(0, 0.1, 0.2, 0.3,
0.4, 0.5, 1)
give an answer in the case below the threshold. Ta-
ble 6 shows some real examples for 3-bond proteins
whose prediction can be corrected by the pattern-wise
BKS method, while the original predictions made by
the classifiers are wrong.
However, the pattern-wise BKS method is not
suitable for the prediction of every protein. The num-
ber of all possible combinations of patterns grows
rapidly with respect to the number of bonds, so
the number of the training samples is relatively not
enough. We then adopt the pair-wise BKS method
for the prediction of proteins with 4 or 5 bonds. The
pair-wise BKS table records the numbers of the truly
bonded pairs and non-bonded pairs in various prob-
ability intervals from two classifiers, CP
1
F
521
and
CP
2
Label
2
. Table 7 shows an example of the partial
pair-wise BKS table for 5-bond proteins. For every
cysteine pair, we advisably adjust the original prob-
ability from CP
1
F
521
method according to the ratio
of the truly bonded pairs in the pair-wise BKS table.
As the experimental results show in the next section,
we get better prediction accuracies if we adopt differ-
ent methods to solve the DCP problem with different
numbers of bonds.
3.4 The Hybrid Method
Instead of large amount of features used by the SVM
method, Zhao et al. (East Zhao et al., 2005) adopted
only one feature, CSP (cysteine separations profile),
to achieve nearly 50% accuracy in the insufficient
dataset. The CSP of protein x with 2n oxidized cys-
teines (n disulfide bonds) is defined as
CSP
x
= (δ1, δ
2
, . . . , δ
2n−1
)
= (ρ
2
− ρ
1
, ρ
3
− ρ
2
, . . . , ρ
2n
− ρ
2n−1
) (2)
where ρ
i
denotes the sequence position of the ith oxi-
dized cysteine in the protein and δ
i
denotes the sepa-
ration distance between oxidized cysteines i and i+ 1.
The divergence (D) of two CSPs for two proteins
x and y is defined (East Zhao et al., 2005) as follows:
D =
i=2n−1
∑
i=1
|δ
x,i
− δ
y,i
|. (3)
It shows that the CSP is an important global fea-
ture for the DCP problem. Thus, we also combine
the CSP method to our hybrid method. Our hybrid
method for predicting the disulfide connectivity pat-
tern is described as follows.
Algorithm: Hybrid method for DCP.
Input: A protein sequence and the bonding states of
all cysteines in it.
Output: The predicted disulfide connectivity pat-
tern.
Case 1: For a 2-bond or 3-bond protein.
• Step 1.1: If the query meets the threshold in the
pattern-wise BKS method for fusing the results
of CP
1
F
521
and CP
1
F
623
, report this pattern as
the predicted pattern.
• Step 1.2: If the minimum divergence obtained
by the CSP search is less than or equal to the
threshold, report this pattern as the predicted
pattern.
• Step 1.3: For the remaining, take the original
maximum weighted pattern from the CP
1
F
521
method as the predicted result.
Case 2: For a 4-bond or 5-bond protein.
• Step 2.1: If the minimum divergence obtained
by the CSP search is less than or equal to the
threshold, report this pattern as the predicted
pattern.
• Step 2.2: Apply the pair-wise BKS method to
fusing the results of CP
1
F
521
and CP
2
Label
2
.
And then report the answer.
4 EXPERIMENTAL RESULTS
In this section, we present the dataset used in our ex-
periments and performance evaluation criteria of the
DCP problem. We aslo show the experimental results.
4.1 Dataset and Performance
Evaluation
For the fair comparison of the prediction accuracy
with previous works, we use SP39 dataset, which is
the same dataset adopted in some previous works. Ta-
ble 8 illustrates the summary of SP39 dataset. This
dataset was first used by Vullo and Frasconi (Alessan-
dro Vullo and Paolo Frasconi, 2004), and it contains
446 proteins with 2 to 5 disulfide bonds, derived from
the SWISS-PROT release no. 39. We also use the
same way as Wang et al.’s (Chong-Jie Wang et al.,
2012) to divide SP39 dataset into 4 disjoint subsets
TheDisulfideConnectivityPredictionwithSupportVectorMachineandBehaviorKnowledgeSpace
115
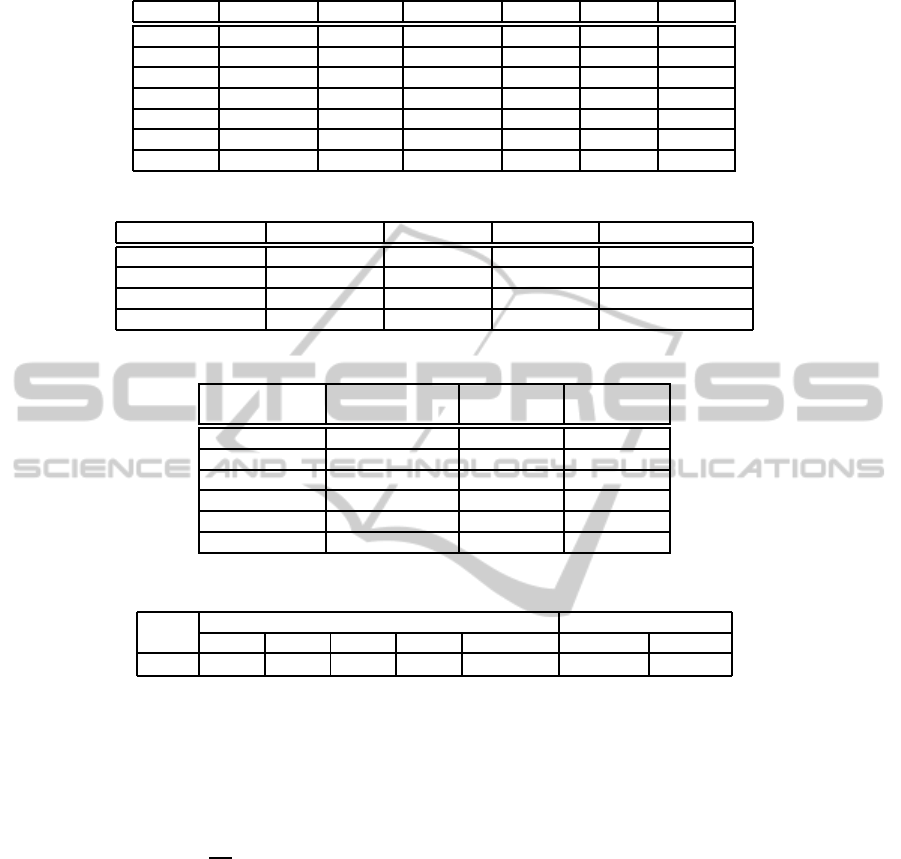
Table 5: An example of the partial pattern-wise BKS table for 2-bond proteins.
CP
1
F
521
Interval CP
1
F
623
Interval 1-1-2-2 1-2-1-2 1-2-2-1
1-1-2-2 (0.15, 0.2) 1-1-2-2 (0, 0.15) 0 1 0
1-1-2-2 (0.15, 0.2) 1-1-2-2 (0.15, 0.2) 5 3 1
1-1-2-2 (0.15, 0.2) 1-1-2-2 (0.2, 0.25) 4 0 0
1-1-2-2 (0.15, 0.2) 1-1-2-2 (0.25, 0.3) 0 0 0
1-1-2-2 (0.15, 0.2) 1-1-2-2 (0.3, 0.35) 0 0 0
1-1-2-2 (0.15, 0.2) 1-1-2-2 (0.35, 0.5) 1 0 0
1-1-2-2 (0.15, 0.2) 1-1-2-2 (0.5, 1) 0 0 0
Table 6: Examples for 3-bond proteins corrected by the pattern-wise BKS method.
Proteins Real patterns CP
1
F
521
CP
1
F
623
Predicted by BKS
CXOA_CONMA 1-2-3-1-2-3 1-2-1-3-2-3 1-2-1-3-2-3 1-2-3-1-2-3
HST1_ECOLI 1-2-3-1-2-3 1-2-1-3-2-3 1-2-1-3-2-3 1-2-3-1-2-3
HCYA_PANIN 1-1-2-2-3-3 1-1-2-2-3-3 1-1-2-3-3-2 1-1-2-2-3-3
CXOB_CONST 1-2-3-1-2-3 1-2-1-3-2-3 1-2-1-3-2-3 1-2-3-1-2-3
Table 7: An example of the partial pair-wise BKS table for 5-bond proteins.
Pairs from
CP
1
F
521
Pairs from
CP
2
Label
2
Truly
bonded
Not
bonded
(0.3, 0.4) (0, 0.1) 0 0
(0.3, 0.4) (0.1, 0.2) 0 1
(0.3, 0.4) (0.2, 0.3) 6 5
(0.3, 0.4) (0.3, 0.4) 4 6
(0.3, 0.4) (0.4, 0.5) 6 6
(0.3, 0.4) (0.5, 1) 1 12
Table 8: The summary of SP39 dataset, where B denotes the number of disulfide bonds.
Number of proteins Number of cysteines
B = 2 B = 3 B = 4 B = 5 B = 2··· 5 Oxidized Total
SP39
*
156 146 99 45 446 2742 4401
*
Defined by Vullo and Frasconi (Alessandro Vullo and Paolo Frasconi, 2004).
for the 4-fold cross-validation. The sequence identity
of proteins between any two subsets is less than 30%.
For the measurement of the performance in con-
nectivity pattern prediction, the accuracy is calculated
as follows:
Q
p
=
C
p
T
p
, (4)
where C
p
denotes the number of proteins whose con-
nectivity patterns are correctly predicted, and T
p
is the
total number of proteins for testing.
4.2 Results
In the CP
1
F
521
method, combined by SVM and the
maximum weighted graph matching (Piero Fariselli
and Rita Casadio, 2001), we find that the prediction
accuracy is very high when the probability of the pre-
dicted pattern is greater than or equal to 0.5 (half).
Thus, before performing our method, the answer is
settled down for these predictions.
Table 9 shows the Q
p
of our methods compared
with previous works in SP39 dataset. The accuracies
of the three SVM models are derivedfrom the patterns
with the maximum weighted graph matching (Piero
Fariselli and Rita Casadio, 2001). However, we find
that it is hard to improve the accuracy by only one sin-
gle SVM model. BKS can play a supporting role in
our method. Although the performance of CP
2
Label
2
is not better than CP
1
F
521
or CP
1
F
623
, CP
2
Label
2
pro-
vides the effect for pair-wise BKS since CP
2
Label
2
represents another concept of pair extraction. As one
can see in the table, with the help of BKS fusing meth-
ods, the accuracy is improved to 65.9%.
Furthermore, when the divergence of CSP is low,
the prediction confidence is also high. Thus, we set
the applicable thresholds of CSP to pick out the pat-
terns as predicted results. Here, we set the threshold
of CSP to 0, 5, 10, and 15 for proteins with 2 to 5
bonds, respectively. Eventually, the prediction accu-
racy of our hybrid method with SVM, BKS and CSP
reaches 69.1%, a great improvement compared with
the previous results.
KDIR2013-InternationalConferenceonKnowledgeDiscoveryandInformationRetrieval
116

Table 9: The Q
p
(in %) of our methods and previous works in SP39 dataset.
Method B = 2 B = 3 B = 4 B = 5 B = 2···5
CSP
a
72.4 54.1 33.3 17.8 52.2
Wang’s method
b
84.0 60.3 55.6 44.4 65.9
CP
1
F
521
84.0 53.4 55.6 46.7 63.9
CP
1
F
623
78.2 60.3 53.5 44.4 63.5
CP
2
Label
2
75.0 49.3 52.5 40.0 58.1
CP
1
F
521
+ BKS 84.0 56.8 55.6 55.6 65.9
CP
1
F
521
+ BKS + CSP 84.0 64.4 57.6 57.8 69.1
a
Proposed by Zhao et al. (East Zhao et al., 2005).
b
Proposed by Wang et al. (Chong-Jie Wang et al., 2012).
5 CONCLUSIONS
According to the study of Wang et al. (Chong-Jie
Wang et al., 2012), which focuses SVM models on
varied features, and the concept of different cysteine-
pair representations proposed by Lu et al. (Chih-Hao
Lu et al., 2007), we do many integrated experiments,
whose results are not all shown in this paper. How-
ever, the improvementof the pure SVM method is not
so significant although the SVM method is still rela-
tively good among the various methods. Some studies
(Bo-Juen Chen et al., 2006; Yu-Ching Chen, 2007)
combine the SVM method with CSP or sequence
alignment to raise the accuracy. The key step of the
CSP method and the sequence alignment method is
to search for a good template set. However, the ac-
curacy of these two methods deeply depends on the
pattern varieties in the template set.
In this paper, we first gather some statistics about
the disulfide bonds, and have successfully found some
rules to discriminate the patterns with high accuracy
in various methods. Then, we adopt the pattern-wise
and pair-wise BKS methods to fuse multiple SVM
models. In addition, the CSP search method is also
invoked in our method. As the experimental results
show, we think that the hybrid method is one of the
good ways to increase the prediction accuracy in the
DCP problem.
In the future, we may apply our hybrid method
to other datasets, and explore more methods for fus-
ing multiple classifiers such as the weighted majority
vote. We may try the CSP method on the inter-bond
template dataset to explore more possibilities of sub-
pattern development.
ACKNOWLEDGEMENTS
This research work was partially supported by the Na-
tional Science Council of Taiwan under contract NSC
100-2221-E-242-003.
REFERENCES
Alessandro Vullo and Paolo Frasconi (2004). Disulfide
connectivity prediction using recursive neural net-
works and evolutionary information. Bioinformatics,
20(5):653–659.
Bo-Juen Chen, Chi-Hung Tsai, Chen-hsiung Chan, and
Cheng-Yan Kao (2006). Disulfide connectivity pre-
diction with 70% accuracy using two-level mod-
els. PROTEINS: Structure, Function, and Genetics,
64:246–252.
Castrense Savojardo, Piero Fariselli, Pier Luigi Martelli,
and Rita Casadio (2013). Prediction of disulfide
connectivity in proteins with machine-learning meth-
ods and correlated mutations. BMC Bioinformatics,
14(S10).
Chao-Chun Chuang, Chun-Yin Chen, Jinn-Moon Yang,
Ping-Chiang Lyu, and Jenn-Kang Hwang (2003). Re-
lationship between protein structures and disulfide-
bonding patterns. PROTEINS: Structure, Function,
and Genetics, 53:1–5.
Chi-Hung Tsai, Bo-Juen Chen, Chen-Hsiung Chan, Hsuan-
Liang Liu, and Cheng-Yan Kao (2005). Improving
disulfide connectivity prediction with sequential dis-
tance between oxidized cysteines. Bioinformatics,
21(24):4416–4419.
Chih-Chung Chang and Chih-Jen Lin (2001). LIBSVM: A
library for support vector machines. Software avail-
able at http://www.csie.ntu.edu.tw/∼cjlin/libsvm.
Chih-Hao Lu, Yu-Ching Chen, Chin-Sheng Yu, and Jenn-
Kang Hwang (2007). Predicting disulfide connectivity
patterns. PROTEINS: Structure, Function, and Genet-
ics, 67:262–270.
Chong-Jie Wang, Chang-Biau Yang, Chiou-Yi Hor, and
Kuo-Tsung Tseng (2012). Disulfide bond prediction
with hybrid models. In Proc. of the 2012 International
Conference on Computing and Security (ICCS ˛a˛e12),
Ulaanbaatar, Mongolia.
David T Jones (1999). Protein secondary structure predic-
tion based on position-specific scoring matrices. Jour-
nal of Molecular Biology, 292(2):195–202.
East Zhao, Hsuan-Liang Liu, Chi-Hung Tsai, Huai-
Kuang Tsai, Chen-Hsiung Chan, and Cheng-Yan Kao
(2005). Cysteine separations profiles on protein se-
quences infer disulfide connectivity. Bioinformatics,
21(8):1415–1420.
TheDisulfideConnectivityPredictionwithSupportVectorMachineandBehaviorKnowledgeSpace
117

F. Ferre and P. Clote (2005). Disulfide connectivity pre-
diction using secondary structure information and
diresidue frequencies. Bioinformatics, 21(10):2336–
2346.
Guantao Chen, Hai Deng, Yufeng Gui, Yi Pan, and Xue
Wang (2006). Cysteine separations profiles on pro-
tein secondary structure infer disulfide connectivity.
In 2006 IEEE International Conference on Granular
Computing, pages 663–665.
Hsuan-Liang Liu and Shih-Chieh Chen (2007). Prediction
of disulfide connectivity in proteins with support vec-
tor machine. Journal of the Chinese Institute of Chem-
ical Engineers, 38(1):63–70.
Jayavardhana Rama G. L., Alistair P. Shilton, Michael M.
Parker, and Marimuthu Palaniswami (2005). Predic-
tion of cystine connectivity using svm. Bioinforma-
tion, 1(2):69–74.
Jianlin Cheng, Hiroto Saigo, and Pierre Baldi (2006).
Large-scale prediction of disulphide bridges using
kernel methods, two-dimensional recursive neural net-
works, and weighted graph matching. PROTEINS:
Structure, Function, and Genetics, 62:617–629.
Leonid A. Mirny and Eugene I. Shakhnovich (1996). How
to derive a protein folding potential? a new approach
to an old problem. Journal of Molecular Biology,
264(5):1164–1179.
Marc Vincent, Andrea Passerini, Matthieu Labbe, and Paolo
Frasconi (2008). A simplified approach to disulfide
connectivity prediction from protein sequences. BMC
Bioinformatics, 9(1):20.
P. Frasconi, A. Passerini, and A. Vullo (2002). A two-stage
svm architecture for predicting the disulfide bonding
state of cysteines. In Proceedings of the IEEE Work-
shop on Neural Networks for Signal Processing, pages
25–34.
Paul M. Harrison and Michael J. E. Sternberg (1994). Anal-
ysis and classification of disulphide connectivity in
proteins : The entropic effect of cross-linkage. Jour-
nal of Molecular Biology, 244(4):448–463.
Pier Luigi Martelli, Piero Fariselli, Luca Malaguti, and Rita
Casadio (2002). Prediction of the disulfide-bonding
state of cysteines in proteins at 88% accuracy. Protein
Science, 11:2735–2739.
Piero Fariselli, Paola Riccobelli, and Rita Casadio (1999).
Role of evolutionary information in predicting the
disulfide-bonding state of cysteine in proteins. PRO-
TEINS: Structure, Function, and Genetics, 36:340–
346.
Piero Fariselli and Rita Casadio (2001). Prediction of
disulfide connectivity in proteins. Bioinformatics,
17(10):957–964.
Pierre Baldi, Jianlin Cheng, and Alessandro Vullo (2005).
Large-scale prediction of disulphide bond connectiv-
ity. In Advances in Neural Information Processing
Systems 17, pages 97–104, Cambridge, MA, USA.
MIT Press.
Rotem Rubinstein and Andras Fiser (2008). Predicting
disulfide bond connectivity in proteins by correlated
mutations analysis. Bioinformatics, 24(4):498–504.
Sarunas Raudys and Fabio Roli (2003). The behavior
knowledge space fusion method: Analysis of general-
ization error and strategies for performance improve-
ment. In In Proc. Int. Workshop on Multiple Classifier
Systems (LNCS 2709, pages 55–64. Springer.
Stephen F. Altschul, Thomas L. Madden, Alejandro A.
Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller,
and David J. Lipman (1997). Gapped blast and psi-
blast: a new generation of protein database search pro-
grams. Nucleic Acids Research, 25(17):3389–3402.
Vladimir N. Vapnik (1999). The Nature of Statistical Learn-
ing Theory. Springer.
Wei-Chun Chung, Chang-Biau Yang, and Chiou-Yi Hor
(2009). An effective tuning method for cysteine state
classification. In Proc. of National Computer Sym-
posium, Workshop on Algorithms and Bioinformatics,
Taipei, Taiwan.
Yu-Ching Chen (2007). Prediction of Disulfide Connectiv-
ity from Protein Sequences. Ph. D. dissertation, Na-
tional Chiao Tung University, Hsinchu, Taiwan.
Yu-Ching Chen and Jenn-Kang Hwang (2005). Pre-
diction of disulfide connectivity from protein se-
quences. PROTEINS: Structure, Function, and Ge-
netics, 61:507–512.
Yu-Ching Chen, Yeong-Shin Lin, Chih-Jen Lin, and Jenn-
Kang Hwang (2004). Prediction of the bonding states
of cysteines using the support vector machines based
on multiple feature vectors and cysteine state se-
quences. PROTEINS: Structure, Function, and Ge-
netics, 55:1036–1042.
KDIR2013-InternationalConferenceonKnowledgeDiscoveryandInformationRetrieval
118
