
Fast and Accurate cDNA Mapping and Splice Site Identification
Micha¨el Vyverman
1
, Dieter De Smedt
1
, Yao-Cheng Lin
2,3
, Lieven Sterck
2,3
, Bernard De Baets
4
,
Veerle Fack
1
and Peter Dawyndt
1
1
Department of Applied Mathematics, Computer Science and Statistics, Ghent University,
Krijgslaan 281 Building S9, B-9000 Ghent, Belgium
2
Department of Plant Systems Biology, VIB, Technologiepark 927, B-9052 Ghent, Belgium
3
Department of Plant Biotechnology and Bioinformatics, Ghent University, Technologiepark 927, B-9052 Ghent, Belgium
4
Department of Mathematical Modelling, Statistics and Bioinformatics, Ghent University,
Coupure links 653, B-9000 Ghent, Belgium
Keywords:
Spliced Alignment, Splice Site Detection, Read Mapping, Long Reads, cDNA Mapping, RNA-seq.
Abstract:
Mapping and alignment of cDNA sequences containing splice sites is an algorithmically and computation-
ally challenging task. Most recently developed spliced aligners are designed for mapping short reads and
sacrifice sensitivity for increased performance. We present mesalina, a highly accurate spliced aligner,
that can also be used to detect novel non-canonical splice sites and whose performance is more robust
with respect to increasing read length. Mesalina utilizes the seed-extend strategy, combining fast retrieval
of maximal exact matches with a sensitive sandwich dynamic programming algorithm. Preliminary re-
sults indicate that mesalina is accurate and very fast, especially for mapping longer reads. In particular,
it is more than ten times faster than mappers with a comparable accuracy. Mesalina is available from
https://github.ugent.be/ComputationalBiology/mesalina.
1 INTRODUCTION
The analysis of the transcriptome is a central part of
biology and requires the analysis of large amounts of
cDNA reads, such as produced by RNA-seq exper-
iments. Evolution in sequencing technology produc-
ing these reads has opened the door for new and larger
experiments, but requires bioinformatics to continu-
ously adapt to larger input datasets and changes in
the features of the reads, including differences in read
length and sequencing errors.
Mapping and alignment of these sequencing reads
against a reference genome is often a first and im-
portant step in the analysis pipeline. In addition to
the computational challenges faced by standard DNA
read mapping, tools for mapping cDNA data from eu-
karyotic genomes have to cope with large gaps in the
alignment caused by introns. Spliced aligners have to
find the exact location of the boundary between in-
trons and exons, called splice sites. In most cases,
splice sites are either surrounded by GT-AG dinu-
cleotides (canonical splice sites) or less frequently
by GC-AG or AT-AC dinucleotides (semi-canonical
splice-sites). In rare cases, however, splice sites are
non-canonical, meaning that they are not surrounded
by any of the previous boundaries.
Depending on the algorithmic strategy employed,
read mappers that deal with spliced alignment can
be divided into two categories (Garber et al., 2011).
Exon-first mappers first align reads without taking
possible splice sites into account. Reads mapped
this way provide a rough map of all the exons of
the reference sequence. The unmapped reads are
split into shorter segments, which are mapped inde-
pendently. Finally, connections between the mapped
segments are searched to identify the exact splice
site locations. Examples of exon-first mappers are
TopHat (Trapnell et al., 2009), TopHat2 (Kim et al.,
2013), MapSplice (Wang et al., 2010), SpliceMap (Au
et al., 2010) and SOAPsplice (Huang et al., 2011).
The second major strategy for spliced alignment is
seed-extend. This approach first filters the reference
sequence to smaller candidate mapping regions us-
ing short matches between read and reference, called
seeds. Seeds can be extended into alignments, can be
chained together to form gapped alignments, or can
233
Vyverman M., De Smedt D., Lin Y., Sterck L., De Baets B., Fack V. and Dawyndt P..
Fast and Accurate cDNA Mapping and Splice Site Identification.
DOI: 10.5220/0004903502330238
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2014), pages 233-238
ISBN: 978-989-758-012-3
Copyright
c
2014 SCITEPRESS (Science and Technology Publications, Lda.)

be used to identify candidate alignment regions that
are explored using more exhaustive methods based
on dynamic programming. This strategy is used,
among others, by GMAP (Wu and Watanabe, 2005),
QPALMA (De Bona et al., 2008), GSNAP (Wu and
Nacu, 2010), and STAR (Dobin et al., 2013). In ad-
dition to the previous categories, spliced aligners are
usually also optimized for a specific type of input
data. Most recent aligners focus on short RNA-seq
reads, whereas GMAP, for example, focuses more on
longer cDNA and EST sequences.
In general, spliced aligners using the seed-extend
approach are several times slower than their com-
petitors using the exon-first strategy. Exon-first ap-
proaches are, however, known to miss spliced align-
ments that also map to the genome contiguously (Gar-
ber et al., 2011). Futhermore, many aligners are de-
signed for mapping very short reads or only allow few
sequence errors between read and genome. Moreover,
many novel spliced aligners are not able to detect rare
non-canonical splice sites. In contrast, mappers that
overcome these shortcomings tend to be much slower
than current short read spliced aligners.
We present mesalina, a seed-extend spliced
aligner that is designed to achieve a high performance
on long spliced reads, while maintaining a high ac-
curacy. The mapper uses techniques from long read
mappers for unspliced reads to speed up the initial
seed finding stage of the algorithm. The extend stage
contains powerful dynamic programming algorithms
introduced by GMAP to achieve high accuracy in de-
tecting the exact splice site locations. Furthermore,
unlike many other novel spliced aligners, mesalina is
also able to detect non-canonical splice sites. Pre-
liminary testing indicates that the current version of
mesalina is more than five times faster than TopHat2
for long reads, while maintaining an accuracy that is
comparable to that of GMAP.
2 METHODS
Mesalina is based on the seed-extend heuristic, which
is widely used among read mappers. The seed-extend
strategy consists of first finding short matches be-
tween read and reference genome using an efficient
index structure. The seeds are utilized to prune the
alignment search space to regions that are close in
size to the length of an alignment. Full alignments
between read and these candidate regions are calcu-
lated in a final extension stage.
Mesalina makes use of maximal exact matches
(MEMs) as seeds of the alignment. For finding
MEMs between read and reference sequences, the
essaMEM program is used (Vyverman et al., 2013).
Subsequently, candidate regions are formed through
clustering of the MEMs. In the final stage of the al-
gorithm, a collinear chain of MEMs forms a gapped
alignment within a candidate region. The gaps be-
tween seeds are filled using dynamic programming.
Gaps spanning an intron are detected using a spe-
cial form of dynamic programming, called sand-
wich dynamic programming, which was introduced
by GMAP (Wu and Watanabe, 2005). The various
stages of the algorithm are discussed in more detail in
the next sections.
2.1 Index
Index structures are commonly used in bioinformat-
ics to speed up searches in large sequence datasets.
This speed-up comes at the cost of a high memory
footprint. As a result, the choice of index structure
can greatly affect the performance of the algorithm.
Most spliced aligners make use of either hash tables
or compressed full-text index structures, such as the
FM-index.
The essaMEM algorithm, incorporated into
mesalina, makes use of an enhanced sparse suffix ar-
ray (ESSA) index structure (Vyverman et al., 2013).
At the base of this index structure lies the suffix ar-
ray index structure (Manber and Myers, 1993), which
stores the lexicographical ordering of all suffixes of
a sequence. Sparse suffix arrays index only one in
s consecutive suffixes, with s the sparseness factor.
The sparseness factor can be set to obtain different
memory-time trade-offs for tools utilizing the index.
Similar to enhanced suffix arrays (Abouelhoda et al.,
2004), sparse suffix arrays can be enhanced with aux-
iliary data structures to simulate traversals on virtual
suffix trees (Vyverman et al., 2013).
2.2 Seed
The first stage in aligning a read is the identification of
subsequence matches between read and reference se-
quences, called seeds. Ideally, seeds should be large
enough to capture as much of the local similarity be-
tween read and reference, but should also be abundant
enough to not miss potential candidate alignment re-
gions.
Mesalina uses maximal exact matches (MEMs)
as seeds. An exact match (s,q,ℓ) between two se-
quences S and Q is a common subsequence of length ℓ
at positions S[s..s+ ℓ] and Q[q..q+ ℓ]. Exact matches
are maximal when the subsequences can not be ex-
tended to the left or right without introducing a mis-
match. In practice, only MEMs of a given minimum
BIOINFORMATICS2014-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
234

length are used, as the number of short (even single
nucleotide) matches that fit the definition is very high,
but less informative than the longer matches.
The essaMEM MEM-finding algorithm is very
fast in practice (Vyverman et al., 2013). In essence,
the algorithm consists of two stages, which are ex-
ecuted for every read suffix. In a first stage, a read
suffix is matched against the ESSA index until a mis-
match occurs, resulting in matches that are right max-
imal. In the second stage of the algorith, left maximal-
ity of a right maximal match is verfied by just compar-
ing the characters preceding the match. In practice,
the algorithm combines the index traversal for several
read suffixes and also contains several techniques to
speed up matching of a single suffix. A detailed de-
scription of the MEM-finding algorithm can be found
in (Vyverman et al., 2013). The minimum MEM-
length can be set by the user and optimal values de-
pend on genome size. Although the use of the ESSA
index limits this minimum length to values larger than
the sparseness of the index, this does not exclude use-
ful values in practice. To further limit the number of
seeds, mesalina can also restrict the set of seeds to the
longest MEMs sharing the same starting position in
the sequencing read.
2.3 Cluster
The set of seeds produced in the previous stage of
the algorithm are divided into clusters of seeds that
are relatively close to each other and form a collinear
chain. Each cluster represents a genomic region in
which the read can have a good alignment.
Currently, mesalina uses a fast greedy chaining
approach. The MEMs are first sorted by reference off-
set, after which the sorted list of MEMs is processed
from left to right. Clusters are formed by consecutive
seeds in the sorted list that (i) are not separated more
than the user-set maximum intron size in the refer-
ence, (ii) do not overlap in the reference and (iii) have
a certain user-set maximum overlap in the read.
For all clusters obtained by the above algorithm,
the percentage of bases in the read that are covered by
seeds in the cluster is calculated. Only clusters with a
high enough coverage percentage are extended. This
filter removes many single-seed clusters and limits the
number of clusters that are extended to only a few in
practice.
2.4 Extend
The extension of a candidate region starts from the
gapped alignment formed by the collinear chain of
seeds contained within the current cluster. In this
gapped alignment, seeds represent long sequences
of matches between read and reference. Gaps be-
tween two consecutive seeds can either result from
differences within the exonic sequence or span an in-
tron. All gaps are resolved using different dynamic
programming routines, similar to the types used in
GMAP (Wu and Watanabe, 2005). The chosen al-
gorithm depends on the difference between the length
of the distance between the two seeds in the reference
sequence, gap
s
, and the gap between the seeds in the
query read gap
q
.
If the difference between gap
s
and gap
q
is smaller
than a given minimum intron length, a basic global
banded alignment is performed over the region de-
fined by the gap between the seeds.
Spliced alignment is performed in the event that
gap
s
− gap
q
is larger than the minimum intron size.
This case is handled using sandwich dynamic pro-
gramming, which was introduced by GMAP (Wu and
Watanabe, 2005) and discussed in detail below.
The converse case, in which gap
q
is far greater
than gap
s
, is also handled by sandwich dynamic pro-
gramming. The extra distance in the read is then cov-
ered by a single long insertion.
Finally, gaps between the seeds at the ends of the
chain and the start/end of the read are handled using
standard semi-global alignment. As a results, no in-
trons can be found that are not surroundedby seeds on
both sides of the intron, which is a known limitation
of the seed-extend strategy.
2.4.1 Sandwich Dynamic Programming
To identify intron boundaries, mesalina uses sand-
wich dynamic programming in a region between two
seeds in a candidate region. Performing a standard
variant of dynamic programming becomes infeasable
in case this region spans an intron, as the intron it-
self can span several thousand nucleotides. In con-
trast, sandwich dynamic programming consists of fill-
ing two smaller dynamic programming matrices and
retrieving splice site locations using a combination of
the scores in both matrices.
The sandwich dynamic programming algorithm is
illustrated in Figure 1. The figure depicts a situation
where two consecutive seeds are separated by a small
gap gap
q
in the read Q and a large gap gap
s
in the
reference sequence S.
The algorithm first performs standard banded dy-
namic programming between the gap
q
region in the
query and two regions of similar size gap
s
′
on the left
and right end of the reference gap. To allow for indels
and some flexibility in alignment, gap
s
′
is a few bases
longer than gap
q
and both gaps include a few bases of
the seeds, as depicted by the small overlap of the gap
FastandAccuratecDNAMappingandSpliceSiteIdentification
235
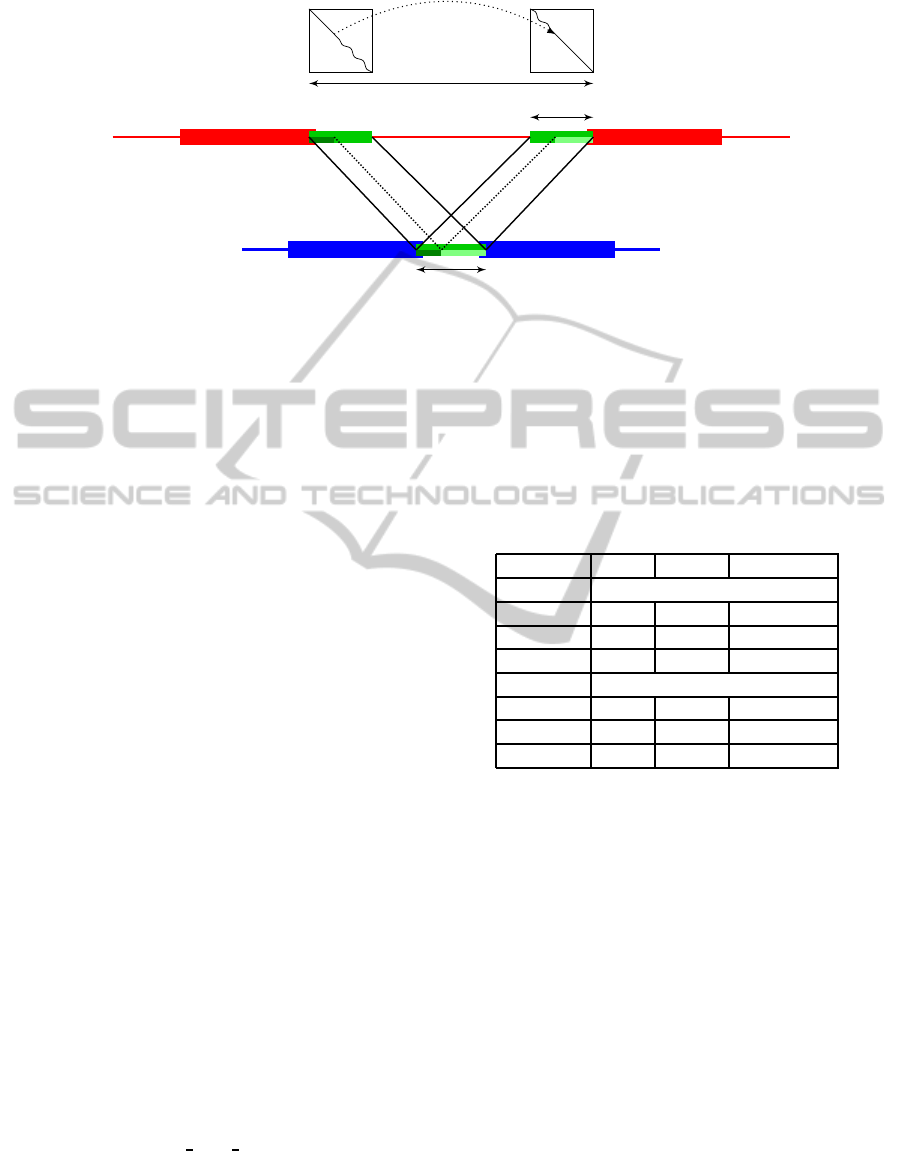
S
Q
gap
s
gap
s
′
gap
q
DP
l
DP
r
Figure 1: Illustration of sandwich dynamic programming between two seeds in a candidate region. The seeds on the reference
sequence S are separated by an intron, whereas the distance gap
q
in the read Q is much smaller. DP
l
and DP
r
represent two
dynamic programming matrices that are computed and have dimension gap
s
′
× gap
q
. Location of the exon-intron boundaries
is decided using a combination of the alignment scores in both matrices. The full alignment consists of traces in DP
l
, DP
r
and the intron gap indicated by the dotted line between the two matrices.
regions and the seeds in Figure 1. Also note that the
matrices DP
l
and DP
r
are filled from opposite corners
due to opposite alignment anchor points.
To find the exact location of the splice site, each
position in gap
q
is tested and receives a score. At the
position with the highest score an intron is inserted.
The score for a position is the sum of three terms:
(i) the maximum score of that position (row) in DP
l
,
(ii) the maximum score of the next position (row) in
DP
r
and (iii) a bonus if the position would result in a
canonical or semi-canonical splice site.
Although this method promotes canonical and
semi-canonical splice sites, it is also able to detect
non-canonical splice sites if no canonical splice sites
are located within the region where dynamic pro-
gramming is performed or if the score for a possible
non-canonical splice site is much higher than possible
canonical splice sites within the same region.
3 RESULTS
Mesalina is written in C++ and is open source (BSD
license). To validate the potential of our approach, we
ran the current implementation of mesalina on sev-
eral simulated read datasets and compared the perfor-
mance and accuracy results against GMAP (Wu and
Watanabe, 2005) (v2013-08-19) and TopHat2 (Kim
et al., 2013) (v2.0.9).
Reads were simulated from Arabidopsis thaliana
(TAIR10, using TAIR10
exon 20101028), using the
RNASeqReadSimulator program (Li, 2012). Three
datasets were produced with varying read lengths of
75bp, 200bp and 500bp. Each dataset contained
100.000 reads with uniform expression profile and
Table 1: Performance and accuracy of spliced aligners on
three read datasets of 100.000 reads, simulated from A.
thaliana. Each column represents a dataset with different
read length. Execution time is measured in seconds and ac-
curacy in percentage of correctly mapped reads.
dataset 75bp 200bp 500bp
run time (s)
mesalina 35 41 52
GMAP 459 849 1532
TopHat2 23 76 240
correctly mapped reads (%)
mesalina 84.4 76.9 62.1
GMAP 85.8 76.1 63.6
TopHat2 83.6 70.1 52.4
simulated substitution errors. An error rate of 5%
was used, which is consistent with PacBio CCS (con-
sensus sequence) data (Roberts et al., 2013). All
tests were run on a single core of a Dell PowerEdge
R610 server with Intel Xeon processor at clock speed
3.07GHz and 48GB RAM running Debian 7.2 and all
tools were run using a single thread and with default
parameter settings.
Test results are summarized in Table 1. Perfor-
mance was measured as the run time of the programs,
excluding index construction time, as this is indepen-
dent of the size of the read data set. Accuracy re-
sults show the percentage of correctly mapped reads.
A read is mapped correctly if the mapper returns an
alignment that maps the read to the correct simulated
mapping position and whose CIGAR-string correctly
identifies the intron boundaries set by the gene anno-
tation data.
Table 1 clearly shows the detrimental impact of
read length on both the accuracy and performance
BIOINFORMATICS2014-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
236

of all tested mappers. This can be explained by an
increased number of reads containing (multiple) in-
trons, especially when reads are longer than the aver-
age exon length (250bp for A. thaliana).
A comparison between mesalina and GMAP is in-
teresting as both seed-extend mappers share several
algorithmic techniques. GMAP is the most accurate
among all tested spliced aligners, but its run time is
much higher than that of the other mappers. Although
mesalina is generally less accurate than GMAP, the
difference in accuracy is relatively small. For reads
of length 200bp, we even report a slightly higher ac-
curacy, although the absolute difference in mapped
reads is small due to the size of the datasets.
TopHat2 is known to be very fast and accurate for
short reads, which is also illustrated by the results of
the 75bp dataset in Table 1. Compared to the other
read mappers, however, its accuracy drops signifi-
cantly for longer reads and its performance drops ten-
fold. Although mesalina is slower than TopHat2 for
shorter reads, it becomes more than four times faster
than TopHat2 for longer reads, while maintaining a
much higher accuracy.
Overall, these preliminary experimental results in-
dicate that our approach achieves a new and inter-
esting performance-accuracy trade-off, especially for
longer reads.
4 DISCUSSION
Many novel spliced aligners are very fast and accurate
for mapping short RNA-seq reads. They are, how-
ever, not designed to handle longer reads and few are
able to detect non-canonical splice sites. In contrast,
mappers designed to map ESTs and longer cDNA se-
quences have a much lower throughput than current
short read mappers. Our goal was to bridge this gap
by combining techniques from long DNA read map-
ping algorithms and sensitive alignment procedures
from GMAP in a novel seed-extend spliced aligner
mesalina.
From an algorithmic perspective, mesalina
demonstrates a promising combination of tried-and-
tested techniques. As a result, the algorithm can either
be seen as a speed-boost for seed-extend algorithms,
such as GMAP, or as technique to provide spliced
alignment support to long read mappers. To the best
of our knowledge, the only algorithm containing a
similar combination of techniques is part of recent
versions of the segemehl read mapper (Hoffmann
et al., 2009). This algorithm uses a combination of
an enhanced suffix array for near-exact matching,
seed chaining and split alignment, which is similar to
sandwich dynamic programming.
The index used by the algorithm for the seed-
finding stage is an enhanced sparse suffix array. This
index structure is related to other suffix array index
structures, but the use of this variant in a spliced
aligner is novel. This index structure requires 9n/s+
n bytes of memory, with n the length of the reference
genome and s the sparseness factor. Although this
is still high compared to other full-text index struc-
tures (Vyverman et al., 2012), the constant term n
could further be lowered by 2 bit encoding the refer-
ence sequence. Furthermore, the memory-time trade-
off of the seed-finding stage can be tuned by chang-
ing the sparseness factor (Vyverman et al., 2013).
In practice, mesalina requires 1.2GB of memory for
s = 1, and only 250MB for s = 9, which is lower
than the index size of GMAP, but higher than that of
TopHat2. Unlike TopHat2, the Memory consumption
of mesalina is independent of the size of the read data
set. It is, however, limited to reference genomes of 4
gigabases due to the use of pointers of 32-bit to posi-
tions in the genome.
The ESSA index structure also allows fast finding
of maximal exact matches (Vyverman et al., 2013).
MEMs are variable length seeds that can contain more
information than short fixed-length seeds and have
already successfully been used in long read map-
ping (Liu and Schmidt, 2012). Based on the seeds
found, candidate alignment regions are identified us-
ing a greedy clustering algorithm and extension of
those regions is determined by the percentage of the
read covered by seeds. Although greedy, this ap-
proach is usually capable of finding the correct map-
ping location. The algorithm still could be improved
by, for example, exploring more than a single al-
ingment per candidate region. Finally, the chain of
seeds is extended into a full alignment using a similar
sandwich dynamic programming strategy as used by
GMAP, although somewhat simplified.
Preliminary experimental results indicate that
mesalina attains the goals that were set and achieves
a new and interesting trade-off between performance
and accuracy. It is much faster than GMAP in all
test cases, while being only slightly less accurate.
It is, however, much more accurate than TopHat2.
Although TopHat2 remains faster for shorter reads,
mesalina performs better for longer reads. We should
remark that these tests are still preliminary and per-
formed on a small dataset. Furthermore, the low ac-
curacy of TopHat2 could be alleviated by tuning com-
mand line parameters.
Although the current version of mesalina already
shows promising results, the algorithm can still be im-
proved to obtain a higher accuracy for reads that are
FastandAccuratecDNAMappingandSpliceSiteIdentification
237

more difficult to map and the implementation could
still be improvedto obtain higher overall performance
and a lower memory footprint.
Within a candidate region, a gapped alignment is
first build using a chain of the seeds found within
this region. The greedy chaining algorithm is cur-
rently the major source of miss-alignments and could
be replaced by an optimal collinear chaining algo-
rithm (Abouelhoda, 2007). Other causes of misalign-
ments include failure to detect splice sites at the ends
of reads and failure to differentiate two consecutive
introns separated by an exon smaller than the mini-
mum seed length.
The run time could be further decreased by select-
ing good settings for parameters, such as minimum
seed length, but also by, for example, using a bit-
parallel dynamic programming implementation in the
extension stage. The memory footprint of the index
could further be reduced by bit-encoding the refer-
ence sequence.
In addition to algorithmic improvements, more
rigorous tests need to be performed on large and var-
ied data sets and experimental results need to be com-
pared to a larger set of spliced aligners, using different
parameter settings.
Finally, the current implementation of mesalina
still lacks some of the features other spliced align-
ers support, including specific algorithms for the de-
tection of micro-exons and alternative splicing, and
paired-end read mapping. We also acknowledge the
need for clear and intuitive command line options and
good portability of the tool.
ACKNOWLEDGEMENTS
The work of MV is supported by the Agency for In-
novation by Science and Technology of the Flemish
government [contract SB-101609]. All authors ac-
knowledge the support of Ghent University: MRP
Bioinformatics: from nucleotides to networks (N2N).
REFERENCES
Abouelhoda, M. (2007). A chaining algorithm for
mapping cDNA sequences to multiple genomic se-
quences. In SPIRE07, 14th international confer-
ence on String Processing and Information Retrieval.
Springer-Verlag.
Abouelhoda, M., Kurtz, S., and Ohlebusch, E. (2004). Re-
placing suffix trees with enhanced suffix arrays. Jour-
nal of Discrete Algorithms, 2:53–86.
Au, K., Jiang, H., Lin, L., Xing, Y., and Wong, W. (2010).
Detection of splice junctions from paired-end RNA-
Seq data by SpliceMap. Nucleic Acids Research,
38:4570–4578.
De Bona, F., Ossowski, S., Schneeberger, K., and R¨atsch, G.
(2008). Optimal spliced alignments of short sequence
reads. BMC Bioinformatics, 9:i170–i180.
Dobin, A., Davis, C., Schlesinger, F., Drenkow, J., Zaleski,
C., Jha, S., Batut, P., Chaisson, M., and Gingeras, T.
(2013). STAR: ultrafast universal RNA-Seq aligner.
Bioinformatics, 29:15–21.
Garber, M., Grabherr, M., Guttman, M., and Trapnell, C.
(2011). Computational methods for transcriptome an-
notation and quantification using RNA-Seq. Nature
methods, 8:469–477.
Hoffmann, S., Otto, C., Kurtz, S., Sharma, C., Khaitovich,
P., Vogel, J., Stadler, P., and Hackerm¨uller, J. (2009).
Fast mapping of short sequences with mismatches, in-
sertions and deletions using index structures. PLoS
Computational Biology, 9:e1000502.
Huang, S., Zhang, J., Li, R., Zhang, W., He, Z., Lam, T.,
Peng, Z., and Yiu, S. (2011). SOAPsplice: genome-
wide ab initio detection of splice junctions from RNA-
Seq data. Frontiers in genetics, 2.
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R.,
and Salzberg, S. (2013). TopHat2: accurate alignment
of transcriptomes in the presence of insertions, dele-
tions and gene fusions. Genome Biology, 14:R36.
Li, W. (2012). RNASeqReadSimulator:
A Simple RNA-Seq Read Simulator.
http://alumni.cs.ucr.edu/ liw/rnaseqreadsimulator.html.
Liu, Y. and Schmidt, B. (2012). Long read alignment
based on maximal exact match seeds. Bioinformatics,
28:i318–i324.
Manber, U. and Myers, G. (1993). Suffix arrays: a new
method for on-line string searches. SIAM Journal on
Computing, 22:935–948.
Roberts, R., Carneiro, M., and Schatz, M. (2013). The
advantages of SMRT sequencing. Genome Biology,
14:405.
Trapnell, C., Pachter, L., and Salzberg, S. (2009). TopHat:
discovering splice junctions with RNA-Seq. Bioinfor-
matics, 25:1105–1111.
Vyverman, M., De Baets, B., Fack, V., and Dawyndt, P.
(2012). Prospects and limitations of full-text index
structures in genome analysis. Nucleic Acids Re-
search, 40:6993–7015.
Vyverman, M., De Baets, B., Fack, V., and Dawyndt, P.
(2013). essaMEM: finding maximal exact matches
using enhanced sparse suffix arrays. Bioinformatics,
29:802–804.
Wang, K., Singh, D., Zeng, Z., Coleman, S., Huang, Y.,
Savich, G., He, X., Mieczkowski, P., Grimm, S., and
Perou, C. (2010). MapSplice: accurate mapping of
RNA-Seq reads for splice junction discovery. Nucleic
Acids Research, 38:e178–e178.
Wu, T. and Nacu, S. (2010). Fast and SNP-tolerant detec-
tion of complex variants and splicing in short reads.
Bioinformatics, 26:873–881.
Wu, T. and Watanabe, C. (2005). GMAP: a genomic map-
ping and alignment program for mRNA and EST se-
quences. Bioinformatics, 21:1859–1875.
BIOINFORMATICS2014-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
238
