
Genome Sequences as Media Files
Towards Effective, Efficient, and functional Compression of Genomic Data
Tom Paridaens
1
, Wesley De Neve
1,2
, Peter Lambert
1
and Rik Van de Walle
1
1
Multimedia Lab, Ghent University - iMinds, Gaston Crommenlaan 8 bus 201, 9050 Ghent, Belgium
2
Image and Video Systems Lab, KAIST, Daehak-ro 291, Guseong-dong, Yuseong-gu, Daejeon 305-701, South Korea
1 STAGE OF THE RESEARCH
In the past decade, the cost of sequencing a human
genome has evolved from millions of dollars to
a few thousands of dollars. As a result of this
exponential drop in cost, the amount of sequencing
data is increasing exponentially (Guy Cochrane et al.,
2013)(Yuichi Kodama et al., 2011). Doubling rates
reach down to four months. As storage capacity per
dollar doubles roughly every 18 months, the storage
of genomic data is becoming a problem.
Given the above observations, a substantial amount
of research is done on the compression of genomic
data. However, there is no such thing as genomic
data from a compression point-of-view. Depending
on the type of species (e.g., genomes of humans, bac-
teria, viruses, and tumors), genomic data possesses
different characteristics. Many compression tools are
optimized for a specific type of genomic data or, at
least, perform differently depending on the type of
species.
Furthermore, the type of storage (stand-alone files
or databases) offers possibilities for different types
of compression (e.g., stand-alone versus reference-
based compression).
Effectivity (compression ratio) is only one aspect of
managing genomic data. Next to effectivity, there
is also a need for efficient management, efficient
transmission over networks and efficient analysis of
the data. To our knowledge, these aspects are often
neglected.
1.1 Targeted Use Case
A group of scientists wants to create a database con-
taining genomic sequences of different species. For
later reference, the reads of the sequencing machines
also need to be stored, including the quality measure-
ments. This database will be used both locally and on
remote sites. Local access to the database is mainly
done in an ad hoc way at the genomics level (Single
Nucleotide Polymorphism or SNP detection, identifi-
cation). The speed of availability (the time between
sequencing and downloading it from the database)
is more important than the compression rates. Re-
mote access is mainly performed for metagenomics
research and runs over a standard internet connection.
The compression ratio, network load and ability to
compare genomic data at the lowest complexity are
key here. As most of the sequences are confidential,
the scientists also have the responsibility to protect the
privacy of their patients. Therefore, encryption of the
data and Digital Rights Management (DRM) are not
to be neglected.
2 OUTLINE OF OBJECTIVES
Compression algorithms can be mapped on a triangle
with three axes (Figure 1):
• Effectiveness: what is the compression ratio of-
fered by the algorithm?
• Efficiency: what is the computing time to com-
press data? What is the transmission efficiency?
• Functionality: does the algorithm allow for added
functionality (e.g. random access, metadata, error
correction,...)?
Functionality
Effectiveness
Efficiency
Figure 1: The three axes of compression algorithms.
3
Paridaens T., De Neve W., Lambert P. and Van De Walle R..
Genome Sequences as Media Files - Towards Effective, Efficient, and functional Compression of Genomic Data.
Copyright
c
2014 SCITEPRESS (Science and Technology Publications, Lda.)

In our research, we will mainly focus on function-
ality and efficiency.
Good compression (effectiveness) is important but
might be inefficient in a network context. In gen-
eral, full-file compression offers higher compression
ratios than block-based compression. However, full-
file compression requires the transmission of the com-
plete files, even when only a small part of the data is
needed. As a result, a lot of data is transmitted with-
out ever being used, causing excessive and unneces-
sary network load.
To maximise efficiency, a novel compression algo-
rithm will be designed that splits up the data into
small blocks. Each of these blocks will be encoded
separately using prediction tools. These tools will
predict blocks either with a reference (a previously
encoded block) or without a reference. This block-
based approach is expected to affect the effectiveness
of the compression algorithm but will offer higher ef-
ficiency and more functionality.
For this block-based compression scheme we will in-
vestigate features such as:
• Random Access, to allow transmission of partial
data;
• Streaming, to offer live access to sequencing
data;
• Parallel Processing, to speed up compression and
decompression;
• Metadata, to facilitate the management of data
and to support random access;
• Encryption, to protect privacy;
• Editing and Analysis in the Compressed Do-
main;
• Error Detection and Correction, to ensure that
the data transmission is error free;
• Load-balancing, choosing a subset of prediction
tools to balance computing time, compression ra-
tio and network load;
• Smart Prediction Tool Selection, to optimise
compression efficiency by selecting a subset
of compression tools based on the different
characteristics of different species genomes.
Despite the focus on efficiency and functionality, it is
clear that effectiveness is not to be neglected. During
our initial research, we discovered that there is no ref-
erence benchmark set to compare algorithms (Sebas-
tian Wandelt et al., 2013). It is therefore a side goal
to define such a reference benchmark set that can be
shared amongst researchers to compare different solu-
tions in speed, memory usage and compression ratio.
In order to be accepted generally, the benchmark set
will need to consist of several types of genomic data:
• Reads (with quality information) and full se-
quences;
• Aligned reads and unaligned reads;
• Genes, chromosomes and full genomes;
• Bacteria, viruses, humans and other species.
Note that the ideal genomic data compression tool of-
fers support for all these types of genomic data.
3 RESEARCH PROBLEM
In this doctoral research, we aim at answering the
following overall research question:
“How can we apply existing technologies in me-
dia compression, storage, management and analysis
to genomic data in such a way that they improve on
current existing technologies in the sense of compres-
sion, transmission and/or analysis efficiency?”
The above research question can be broken up
into a number of smaller research questions:
“How can we design a file format for the compres-
sion of genomic data in such a way that it supports all
features that are currently already available for, e.g.,
video files? These features include, amongst others,
random access, streaming capabilities, metadata, en-
cryption, DRM, load-balancing, compressed-domain
manipulation, parallel processing, error detection
and error correction.”
“Which set of prediction and coding tools pro-
vides the highest compression ratio?”
“How does random access affect the compression
efficiency and network load?”
“How does error correction affect the compres-
sion efficiency? Does it consume the compression
gain?”
“Which set of prediction and coding tools pro-
vides the highest compression ratio? Can these tools
compensate for the loss of efficiency due to the use of
block-based compression?”
“How can we integrate the proposed file format
into existing technologies that allow re-use of already
existing file streaming technologies?”
BIOSTEC2014-DoctoralConsortium
4

“How can we compare the performance of our
solution with already existing solutions in terms of
compression ratio, (de)compression time and network
load?”
4 STATE OF THE ART
4.1 Compression of Genome Sequences
Genome compression is split up in two different dis-
ciplines: full-sequence compression and read com-
pression. Both disciplines cover the compression of
plain sequencing data (a complete sequence or sepa-
rate reads). The latter will also support the compres-
sion of quality information. To compress the genomic
data, four different types of compression are avail-
able(Sebastian Wandelt et al., 2013):
1. Bit Encoding - Bases are stored as a sequence of
two bits (or three bits when the N base is also
used), instead of the eight bits needed for en-
coding the sequence in the ASCII-format. This
type of encoding offers a 4:1 compression ratio
(or 2.67:1 for sequences that use the N base).
2. Dictionary-based Encoding - Groups of bases
are stored as a reference to previously encoded
parts of the sequence(Shanika Kuruppu et al.,
2011)(Xin Chen et al., 1999).
3. Statistical Encoding - Bases are predicted, based
on a probabilistic model. If these predictions are
reliable, statistics encoding can be highly effi-
cient(Armando J. Pinho et al., 2009).
4. Reference-based Encoding - Groups of bases
are stored as a reference to another reference
genome. As genomes are highly similar be-
tween species of the same kind, these compres-
sion schemes can reach compression ratios of
more than 100:1(Markus Hsi-Yang Fritz et al.,
2011).
4.2 Media files
In the world of media file
1
compression and transmis-
sion, a substantial amount of research has been (and
is still being) performed on effectivity, efficiency and
functionality.
Organisations such as MPEG (Moving Picture Ex-
perts Group) and JCT-VC (Joint Collaborative Team
on Video Coding) define technical specifications for
1
A file containing video content, audio content and/or
pictures
video and audio compression. These technical spec-
ifications focus mainly on effectivity (and efficiency
to some extent). Next to these compression formats,
MPEG and JCT-VC define technical specifications to
add functionality such as media containers for trans-
portation and metadata standards.
MPEG and JCT-VC update their video and audio
compression specifications on a regular basis. This
is necessary to cope with the ever growing amount
of bandwidth and storage capacity that is needed for
storing video and other media files. The amount of,
e.g., video data created every day is rising exponen-
tially. While filming was limited to dedicated devices
until a few years ago, now nearly every computer,
smartphone and tablet can generate Full HD (High
Definition) videos (or even videos at higher resolu-
tions).
Smartphones and tablets are for media what Next
Generation Sequencing (NGS) is for genomes: a
cheaper way to create data and, as a result, the source
of an exponential growth in data.
Thanks to the standardisation of these compression
formats, researchers can focus on more advanced re-
search, compare their results and built on research of
colleagues.
These standardisation efforts opened doors for re-
search on many surrounding topics:
• Metadata;
• Compressed-domain editing, adaptation and
detection;
– H.264 AVC (Advanced Video Coding)(Chris
Poppe et al., 2009);
– H.264 SVC (Scalable Video Coding)(Heiko
Schwarz et al., 2007);
– H.265 HESVC (High-Efficiency Scalable
Video Coding);
– MPEG-21 BSDL, a format-agnostic editing
tool(Tom Paridaens et al., 2007);
• Speed improvements;
– transcoding (Fatma Al-Abri et al., );
– parallel processing;
– GPU (Graphical Processing Unit) accelera-
tion(Pie, );
• Streaming;
– servers;
– streaming formats;
– adaptive streaming(Davy Van Deursen et al.,
2010);
• Adaptivity;
– definition of levels (levels define e.g. computa-
tional complexity);
GenomeSequencesasMediaFiles-TowardsEffective,Efficient,andfunctionalCompressionofGenomicData
5

– customisable compression tools;
– balancing compression rate versus processing
power/energy consumption;
• Encryption(Glenn Van Wallendael et al., 2013);
• Random access.
Many of these topics can be introduced in the
context of genomic data, provided the compression
scheme supports features such as random access and
metadata.
4.3 MPEG-21 BSDL:
Compressed-domain Editing and
Adaptation
In previous research by the authors, it was demon-
strated that the MPEG-21 BSDL (Bitstream Syntax
Description Language) standard can be used to ef-
ficiently adapt existing media files before stream-
ing them to end-users. MPEG-21 BSDL is format-
agnostic and allows fast adaptation of files. As only
limited processing has to be performed, files do not
have to be decoded to select pieces of that file (Davy
Van Deursen et al., 2010). As MPEG-21 BSDL is
format agnostic, it is also applicable to any other file
format, including our proposed solution as long as a
Bitstream Syntax (BS) Scheme is available for that
file format. This scheme describes the binary syntax
of the file format.
The description and adaptation of a data file happens
in three steps:
1. The generation of a Bitstream Syntax Description
(BSD). This is an eXtensible Markup Language
(XML) file that contains the structure of the media
file. To generate this file, a BS Scheme is used.
This step only needs to be performed once per file.
The resulting BSD can be reused for any future
adaptation on that file.
2. The BSD file is adapted. This can be done manu-
ally or using standard XML transformation tech-
niques. Typical adaptations are the selection of,
e.g., a movie scene. To do so, the data corre-
sponding to all other scenes in that video scene
are removed from the description.
3. Based on the adapted BSD file, the adapted media
file is generated.
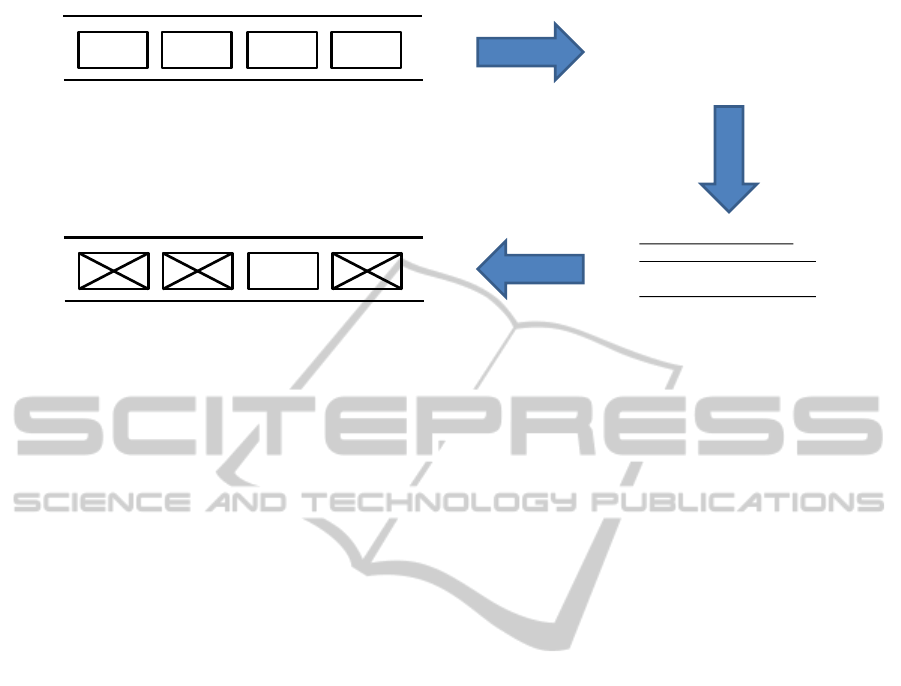
Figure 2 illustrates this process, applied to ge-
nomic data. In Step 1 (generation), the description
of a sequence of 4 genes is created. In this example,
the genes are defined with their respective first and
last bit. As an example, we will select Gene3.
In Step 2 (transformation), the description is adapted
by removing all unwanted genes from the description.
In Step 3 (adaptation), the new genomic data file is
created by adapting the original file, in casu deleting
the bits corresponding to the other genes.
4.4 Management and Analysis of
Compressed Genome Sequences
Most genome compression tools are designed to of-
fer maximum compression. To reach maximum com-
pression, functionality is often very limited. Exam-
ples of functionality that many genome compression
formats do not support are:
• Random Access - to access specific parts of files
without full decompression (Kalyan Kumar Kaipa
et al., 2010);
• Streaming - to access genomic data, even during
compression and sequencing;
• Metadata - to allow to integrate additional infor-
mation on the genomic data.
• Encryption - to protect the privacy or information
of delicate subjects;
• Compression Parameters - to enable balancing
of (de)compression speed, memory usage, and
compression ratio;
• Compressed-domain Editing and Analysis - to
edit and analyse without decompressing the data.
• Reads and Full Sequences - to support compres-
sion of both reads (and quality information) and
complete sequence files;
• Parallel (de)Compression - to improve the com-
pression and decompression efficiency.
As a result, most of the genomic data is still stored
non-compressed in order to allow for easy editing
and analysis. There are trials ongoing, e.g., ENA
(European Nucleotide Archive) is adding support for
CRAM, but these only limit data traffic and lack func-
tionality(EBI, 2013).
A number of the features mentioned above can be
applied to existing formats by leveraging additional
tools. Other features, such as parallel compression,
might be harder to implement, or even create no effi-
ciency gain at all, depending on the compression al-
gorithm.
5 METHODOLOGY
We will split up the development of the novel file for-
mat and the corresponding tools into several steps:
BIOSTEC2014-DoctoralConsortium
6
<Genome>
<Gene1>0 1500</Gene1>
<Gene2>1501 3500</Gene2>
<Gene3>3501 5488</Gene3>
<Gene4>5489 8000</Gene4>
</Genome>
Gene1 Gene2 Gene3 Gene4
Step 1
<Genome>
<Gene1>0 1500</Gene1>
<Gene2>1501 3500</Gene2>
<Gene3>3501 5488</Gene3>
<Gene4>5489 8000</Gene4>
</Genome>
Step 2
Gene1 Gene2 Gene3 Gene4
Step 3
Figure 2: Selecting Gene3 from a genomic data file.
• The development of a modular compression archi-
tecture. This architecture will consist of modules
responsible for input, compression, output (multi-
plexing), statistics and metadata.
• The creation of a BSDL scheme (or equivalent),
based on the final data format.
• The implementation and demonstration of the ba-
sic editing functionality in the compressed do-
main, based on the BSDL scheme.
• The integration of the BSDL scheme and the edit-
ing functionality in an existing BSDL-based file
parser and streaming server.
• The integration of the compressed data streams
into existing streaming file formats to enable
use of mainstream (fully-optimised) streaming
servers as backbone servers.
The input module will support:
• Plain ASCII sequence data files;
• FASTA files;
• SAM (Sequence Alignment/Map) files.
The compression module will contain multiple pre-
diction and coding tools based on bit encoding,
dictionary-based encoding and statistical encoding.
The output module will produce binary data, encoded
using CABAC (Context-Adaptive Binary Arithmetic
Coding).
The statistics module will provide users with statis-
tical data on the usage of the different prediction and
coding tools and their compression and computational
performance. The metadata module will handle the
metadata of the input file and provide the list of ran-
dom access points.
Based on this research, additional research topics
are:
• Support for other popular file formats (e.g., the
CRAM specification);
• Data encryption;
• Error protection and correction;
• Compression acceleration (multi-threading, GPU
acceleration);
• Co-creation of a benchmark data set for DNA data
compression in general.
6 EXPECTED OUTCOME
This research will result in a novel genomic data file
format that supports:
• Competitive compression and computational per-
formance (in bits/base and msec/base);
• Random access;
• Error protection;
• Data protection (encryption);
• DRM;
• Metadata;
• Configurable and adaptive encoding.
To test the compression performance, a list of
frequently-used test sequences will be used. We will
attempt to define a reference benchmark data set to-
gether with other researchers and institutes to offer a
reliable comparison of the performance between for-
mats.
Based on this genomic data file format, we will
develop a management and streaming solution that
optimises the needed storage capacity, the network
load and the speed of access to both full genomic se-
quences as well as partial sequences.
GenomeSequencesasMediaFiles-TowardsEffective,Efficient,andfunctionalCompressionofGenomicData
7

7 PRELIMINARY RESULTS
We have implemented a file compression tool that will
serve as a reference for further test results. It is the
base line to which the effect of additional compres-
sion tools and added functionality will be measured.
The tool performs the following steps:
1. The input sequence is split up in blocks of an ar-
bitrary size (currently 48 bases).
2. For each of the bocks, a prediction is made using
six separate tools:
(a) INTRA Coding - the prediction is a repetition
of a single base. E.g., ”AAAAA...”.
(b) INTRA Coding - the prediction is an alter-
nating repetition of two different bases. E.g.,
”ATATAT...”.
(c) HUFFMAN Coding - the input block is coded
with a 2-base Huffman coding. In this case, nu-
cleotides are encoded in pairs. It is expected
that codon-wise encoding would be more effi-
cient.
(d) SEARCH Coding - the prediction is the block
that contains the least differences when com-
pared to the input block. To find this block, the
input block is compared to all previously en-
coded blocks within a range Search Window.
(e) SEARCH INVERSE COMPLEMENT Cod-
ing - the predicion is the same as SEARCH
coding but instead of the block, the inverse
complement of the input block is used during
the search.
(f) HIERARCHICAL Coding - the input block is
being split in half. Each of the parts will then
be encoded with the tools in this list. To avoid
endless splitting, a maximum depth is defined.
3. The differences between a block and its prediction
(residue) are encoded as a sequence of address-
transformation couples. The addresses are en-
coded in plain bits. The transformation informa-
tion is encoded using Huffman coding. The trans-
formation information is based on a couple of op-
erators (complement, switch). The operator com-
plement returns the complement of the predicted
base:
Complement(A) = T Complement(T ) = A
Complement(C) = G Complement(G) = C
The operator switch returns the equivalent base
from the other base pair
2
:
Switch(A) = C Switch(C) = A
Switch(G) = T Switch(T ) = G
2
base pairs are the couples {A,T} and {C,G}
These operators can be combined:
Complement(Switch(T )) = Complement(G) = C
If none of the operators is used, the result is N:
(T ) = N
4. The best prediction mode is chosen based on
compression efficiency. It is encoded in the
output bit stream, together with the residue (the
difference between the prediction and the actual
input block).
In early tests, we used a sequence of the Y chro-
mosome of a human. This file contains both A,C,G,T
bases and the undefined N base. In Table 1, we
show the compression efficiency of a number of stan-
dard solutions compared to our compression tool. To
store the Y chromosome sequence in ASCII, 8 bits
are needed per base. Even in pure binary format, it
would still take 3 bits per base. When Huffman would
be used, this specific file would need 2.21 bits per
base. Our block-based solution needs, depending on
the configuration, less than 2 bits per base.
Table 1: Compression efficiency.
Encoding bits/base
ASCII 8
Binary (ACGTN) 3
Huffman 2.21
Our solution <2
It is obvious that several improvements can be in-
troduced to further enhance the above algorithm. Pos-
sible improvements are:
• The use of arithmetic coding instead of HUFF-
MAN coding;
• The use of context-adaptive arithmetic coding
based on the different types of encoded data;
• Additional prediction schemes, including support
for single-nucleotide insertions and deletions;
• Optimisation of the encoding of the prediction
signalisation and the resulting residue.
However, the current format already inherently sup-
ports:
• Parallel processing, thanks to the fixed block size
(provided the search windows are managed well);
• Load-balancing, thanks to the search parameters
and enabling/disabling of prediction tools;
• Random access, thanks to the block-based coding.
It should be noted that we can expect that some func-
tionality, such as random access and error protection,
will decrease the compression efficiency of the tool.
BIOSTEC2014-DoctoralConsortium
8

ACKNOWLEDGEMENTS
The research activities described in this paper were
funded by Ghent University, iMinds, the Institute for
the Promotion of Innovation by Science and Tech-
nology in Flanders (IWT), the Fund for Scientific
Research–Flanders (FWO–Flanders), and the Euro-
pean Union.
REFERENCES
Armando J. Pinho et al. (2009). DNA coding using finite-
context models and arithmetic coding. ICASSP.
Chris Poppe et al. (2009). Moving object detection in
the H.264/AVC compressed domain for video surveil-
lance applications. Journal of visual communication
and image representation, 20(6):428–437.
Davy Van Deursen et al. (2010). NinSuna : a fully in-
tegrated platform for format-independent multime-
dia content adaptation and delivery using Semantic
Web technologies. Multimedia tools and applications,
46(2-3):371–398.
EBI (2013). First bulk CRAM submission to ENA.
EBI. http://www.ebi.ac.uk/ena/about/news/first-bulk-
cram-submission-ena.
Fatma Al-Abri et al. Optimal H.264/AVC video transcod-
ing system. Multimedia tools and applications, pages
335–336.
Glenn Van Wallendael et al. (2013). Encryption for high
efficiency video coding with video adaptation capa-
bilities. IEEE international conference on consumer
electronics.
Guy Cochrane et al. (2013). The future of DNA sequence
archiving. Gigascience.
Heiko Schwarz et al. (2007). Overview of the Scalable
Video Coding Extension of the H.264/AVC Standard.
IEEE transactions on circuits and systems for video
technology, 17(9).
Kalyan Kumar Kaipa et al. (2010). System for Random
Access DNA Sequence Compression. IEEE Interna-
tional Conference on Bioinformatics and Biomedicine
Workshops.
Markus Hsi-Yang Fritz et al. (2011). Efficient stor-
age of high throughput DNA sequencing data using
reference-based compression. Cold Spring Harbor
Laboratory Press.
Sebastian Wandelt et al. (2013). Trends in genome com-
pression. Journal of Current Bioinformatics.
Shanika Kuruppu et al. (2011). Optimized Relative Lempel-
Ziv Compression of Genomes. 34th Australasian
Computer Science Conference.
Tom Paridaens et al. (2007). XML-driven Bitrate Adap-
tation of SVC Bitstreams. 8th International Work-
shop on Image Analysis for Multimedia Interactive
Services.
Xin Chen et al. (1999). Compression Algorithm for DNA
Sequences and Its Applications in Genome Compari-
son. Genome Inform Ser Workshop Genome Inform.
Yuichi Kodama et al. (2011). The sequence read archive:
explosive growth of sequencing data. Nucleic Acids
Research.
GenomeSequencesasMediaFiles-TowardsEffective,Efficient,andfunctionalCompressionofGenomicData
9
