
Modeling Genetical Data with Forests of Latent Trees for Applications in
Association Genetics at a Large Scale
Which Clustering Method should Be Chosen?
D.-T. Phan
1
, P. Leray
1
and C. Sinoquet
2
1
LINA / UMR CNRS 6241, Polytech/ University of Nantes, rue Christian Pauc, BP 50609, 44306 Nantes, France
2
LINA / UMR CNRS 6241, Faculty of Sciences, University of Nantes, 2 rue de la Houssini`ere, 44322 Nantes, France
Keywords:
Linkage Disequilibrium, Genome-wide Association Study, Multilocus Association Study, Data Dimension
Reduction, Probabilistic Graphical Model, Bayesian Network.
Abstract:
Association genetics, and in particular genome-wide association studies (GWASs), aim at elucidating the
etiology of complex genetic diseases. In the domain of association genetics, machine learning provides an
appealing alternative framework to standard statistical approaches. Pioneering works (Mourad et al., 2011)
have proposed the forest of latent trees (FLTM) to model genetical data at the genome scale. The FLTM is a
hierarchical Bayesian network with latent variables. A key to FLTM construction is the recursive clustering of
variables, in a bottom up subsuming process. In this paper, we study the impact of the choice of the clustering
method to be plugged in the FLTM learning algorithm, in a GWAS context. Using a real GWAS data set
describing 41400 variables for each of 3004 controls and 2005 individuals affected by Crohn’s disease, we
compare the influence of three clustering methods. Data dimension reduction and ability to split or group
putative causal SNPs in agreement with the underlying biological reality are analyzed. To assess the risk
of missing significant association results through subsumption, we also compare the methods through the
corresponding FLTM-driven GWASs. In the GWAS context and in this framework, the choice of the clustering
method does not impact the satisfying performance of the downstream application, both in power and detection
of false positive associations.
1 INTRODUCTION
With the finalization of the Human Genome Project
in 2003, it was confirmed that any two individuals
share, on average, 99.9% of their genome with one
another. It is then the sole 0.1% genetic variations that
may explain why individuals are physically different
or should inherit a greater risk of contracting genetic
disorders, such as coronary heart disease, diabetes,
autism, some cancers. As a consequence, identify-
ing the genetic factors underlying these diseases po-
tentially plays a crucial role in prediction, monitoring
subjects with risks, as well as developing new treat-
ments. Deciphering the putative causes of complex
genetic diseases has been one of the main focuses of
human genetics research during the last thirty years.
Among different approaches that have been proposed,
association studies stand out as one of the most suc-
cessful paths, even though their potential is yet to be
fully tapped.
The HapMap Project (Gibbs et al., 2003) and
its successor, the 1000 Genomes Project (The 1000
Genomes Project Consortium, 2010), were launched
with the hope to establish a catalogue of human
genome regions in which people of different popula-
tions have differences.
When no clue is available about the genome re-
gions likely to contain one of the putative causes for
a studied disease, geneticists are compelled to resort
to genome-wide association studies (GWASs). Ge-
netic markers are used for this purpose, as well as
a population of affected and unaffected individuals.
Genetic markers represent as many DNA sequences,
spread over the whole genome, with a known loca-
tion, where the DNA variations within a given popu-
lation may be observed. In a nutshell, GWASs seek
to identify genetic markers whose variants vary sys-
tematically between affected (cases) and unaffected
(controls) individuals (Balding, 2006). The standard
GWAS consists in comparing variant frequencies in
cases and controls, on massive genotypic datasets
(tens of thousands of individuals each described by
5
Phan D., Leray P. and Sinoquet C..
Modeling Genetical Data with Forests of Latent Trees for Applications in Association Genetics at a Large Scale - Which Clustering Method should Be
Chosen?.
DOI: 10.5220/0005179800050016
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2015), pages 5-16
ISBN: 978-989-758-070-3
Copyright
c
2015 SCITEPRESS (Science and Technology Publications, Lda.)

hundreds of thousands up to a few millions of ge-
netic markers). The goal is to identify the loci on
the genome for which the distributions of variants are
significantly different between cases and controls, us-
ing dependence - namely association - tests (e.g. the
Chi
2
test). The unit variants, called single nucleotide
polymorphisms (SNPs), which refer to single base
pair changes in the DNA sequence, represent the most
abundant type of variants in human; they are very of-
ten used as markers in GWASs.
The key to GWASs lies in this interesting phe-
nomenon known as ”linkage disequilibrium” (LD)
where variants for different SNPs tend to co-occur
non-randomly (Pritchard and Przeworski, 2001) (the
corresponding SNPs are said to be in LD). The case
would be exceptional if a genetic marker, which is
observed in the population, coincided with a genetic
causal factor. Nevertheless, thanks to LD, a depen-
dence exists between the non observed causal factor
and a genetic marker nearby the former. On the other
hand, by definition, a dependence exists between the
causal factor and the disease of interest. Therefore, it
is likely that a dependence will be detected between
the nearby genetic marker and the disease.
In the human genome, the HapMap project con-
firmed evidence of the linkage disequilibrium, this la-
tent structure organized in the so-called ”haplotype
blocks”. Therein, regions showing high dependences
between contiguous markers (blocks) alternate with
shorter regions characterized by low statistical depen-
dences. In general, LD exhibited among physically
close loci is stronger than LD between SNPs that are
farther apart. In other words, LD decays with dis-
tance.
However, standard GWASs do not fully exploit
LD. Some authors proposed to test combinations of
SNPs - haplotype blocks - against the disease, rather
than merely each SNP against the disease: this is the
principle of multilocus approaches. First, if the causal
SNP has low frequency and is not in high LD with
any one of the genotyped SNPs, then the multilocus
test will tend to be more powerful. Besides, the ad-
vantage to the GWAS is that the LD is likely to reveal
an excess of haplotype sharing around a causative lo-
cus, amongst cases. Third, testing haplotypes instead
of SNPs is a way to implement data dimension re-
duction. In this context, fine LD modeling at genome
scale is required.
Few works have focused on LD modeling at
genome scale, which is a challenging task. The pro-
posals of (Abel and Thomas, 2011) and (Verzilli et al.,
2006) both rely on the use of Markov random fields,
a popular kind of probabilistic graphical models. Two
scalable models designed for the specific purpose of
multilocus GWASs have been described by (Brown-
ing and Browning, 2007) and (Mourad et al., 2011).
The approach in (Browning and Browning, 2007) re-
lies on a variable length Markov chain (VLMC), a
Markov model where the size of the memory con-
ditioning the prediction of the variant at a given lo-
cation is flexible. In contrast with this block-based
method, the works in (Mourad et al., 2011) seek to
subsume clusters of SNPs through latent variables.
SNPs within the same cluster are not necessarily con-
tiguous. Such latent variables are intented to be tested
against the disease. Both methods account for the
fuzzy nature of LD since block boundaries are not ac-
curately defined over the genome. However, being
blocked-based, the method in (Browning and Brown-
ing, 2007) cannot take into account long-range depen-
dences. Moreover, LD is intrinsically hierarchical,
with clusters of SNPs recursively structured in clus-
ters of lower and lower correlated SNPs. To attempt
a faithful representation of LD upstream of a GWAS,
hierarchical clustering is one of the key ingredients of
the learning algorithm of the Bayesian model used in
(Mourad et al., 2011). Since clustering is central to
learning the model in (Mourad et al., 2011), namely
the forest of latent tree models (FLTM), this paper
analyses the impact of the choice of the clustering
method in a GWAS context.
2 OBJECTIVES AND
ORGANIZATION OF THE
PAPER
In the remainder of this paper, data partitioning - or
clustering - denotes the generation of a set of non
overlapping clusters. Such a task is highly complex.
Though, choosing a clustering method to learn an
FLTM must comply with the scalability goal. This
paper compares the native clustering method used in
(Mourad et al., 2011) (CAST
bin
) with a relaxed ver-
sion (CAST
real
) and another clustering method (DB-
SCAN). In this framework, two aims of the paper are
to evaluate whether FLTM learning is robust to the
choice of the clustering method and how close a clus-
tering method approximates the underlying biological
reality. To fulfill the first goal, a protocol is used that
relies on assessing how much two partitions agree.
The second objective is met by applying the previ-
ous protocol to compare each clustering method to a
reference partition supposed to be close to biological
reality. The Haploview software program is the tool
chosen to derive such a reference partition. Focusing
on the data dimension reduction aspect, a third objec-
BIOINFORMATICS2015-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
6

tive of the paper is to analyze the impact of the choice
of the clustering method on data subsumption quality.
By construction, an FLTM-based GWAS processes
data subsumed through latent variables, to hopefully
pinpoint the interesting regions of a genome without
testing each SNP for association. Thus, the third ob-
jective of this paper is to assess whether the choice
of the clustering method impacts the risk of missing
significant association results through subsumption.
FLTM-driven GWASs are run to study this impact.
The remainder of the paper is organized in five
sections. Section 3 first offers a brief introduction to
Bayesian networks, the kind of probabilistic graphi-
cal models FLTM is based upon. Then section 3 pro-
vides a broad brush description of the FLTM learning
algorithm together with a sketch of a GWAS strat-
egy based on FLTM. Section 4 briefly refers to the
native clustering method used in FLTM (CAST
bin
)
and to its relaxed version (CAST
real
); it then moti-
vates the choice of the alternative clustering method
(DBSCAN) plugged in the FLTM learning algorithm.
Then, section 5 explains the design of the protocols
and methods used in our work. First, we discuss
the protocol used to assess how much two partitions
agree. Second, we justify the use of the Haploview
software program to derive the reference partition,
supposedly the closest representation of the underly-
ing reality. In section 6, we describe the Crohn’s dis-
ease GWAS data used in our study. Section 7 is de-
voted to the presentation and discussion of the results
observed.
3 FRAMEWORK AND FLTM
MODEL
The FLTM model is a tree-structured Bayesian net-
work (BN). Therefore this section first briefly in-
troduces Bayesian networks, to further focus on the
FLTM model. The principle of the FLTM learning
algorithm is then presented. Finally, the principle
for a multilocus GWAS based on the FLTM model
is sketched.
3.1 A Brief Reminder about
Probabilistic Graphical Models
When probabilistic graphical models are learnt from
scratch, one has to learn their two fundamental com-
ponents from a data matrix. In this matrix, the lines
correspond to the observations and the columns cor-
respond to the variables X
i
(1 ≤ i ≤ n). For exam-
ple, in the case of genetical data, the observations
(a) (b) (c)
Figure 1: (a) Latent tree model (LTM). (b) Forest of la-
tent tree model (FLTM). (c) Latent class model. Observed
and latent variables are represented in dark and light color
shades, respectively.
are the individuals (cases and controls) in the popu-
lation studied, and the variables are the SNPs. The
qualitative component of a BN is a graph where the
variables are represented as nodes. The connections
between the nodes represent the direct dependences
between the variables. More specifically, the qualita-
tive component of a BN is a directed acyclic graph.
The quantitative component of a BN is a collection of
probability distributions, denoted as ”the parameters
θ”. If the variable X
i
has no parent in the graph, then
θ
i
is merely an a priori distribution (θ
i
= P(X
i
)). If
the variable X
i
has a set of parents Pa
X
i
, then θ
i
is the
conditional distribution θ
i
= P(X
i
| Pa
X
i
). In particu-
lar, Bayesian networks offer a practicable framework:
exploiting the network structure, this framework al-
lows to compute the joint probability of the variables,
P(X), as a product of low-dimensional functions.
It may happen that the data observed is thought to
embed a latent structure, depicted through latent vari-
ables and their connections in the learnt BN. In this
case, learning the Bayesian network encompasses the
task of inferring the latent variables, and their connec-
tions within the BN.
The FLTM model is a forest of latent tree models
(LTMs). The Figure 1(a) shows that an LTM is char-
acterized by a hierarchical structure organized in lay-
ers. The first layer is composed of the observed vari-
ables. The other layers are composed of latent vari-
ables. The learning algorithm of the FLTM (see Fig-
ure 1(b)) relies on the simplest LTM that may be de-
scribed, the latent class model (LCM). A latent class
model connects a single latent variable to child vari-
ables ; no connections are allowed between the latter
(see Figure 1(c)).
3.2 Sketch of the FLTM Learning
Algorithm
Learning a BN is a hard task that consists in inferring
both the graph structure and the parameters. Learn-
ing a BN with latent variables is far more compli-
cated. First, one does not even know how many latent
ModelingGeneticalDatawithForestsofLatentTreesforApplicationsinAssociationGeneticsataLargeScale-Which
ClusteringMethodshouldBeChosen?
7
variables have to be inferred. Second, in a BN with-
out latent variables, the parameters are estimated to
maximize the likelihood, that is the probability of the
(observed) data given the parameters. In contrast to
this rapid algorithm, a slow procedure has to be em-
ployed for BNs with latent variables, the expectation-
maximization algorithmdedicated to learn parameters
in the case of missing data. Prior knowledge (the hi-
erarchical LD structure) is used by the specific pro-
cedure described in (Mourad et al., 2011), to provide
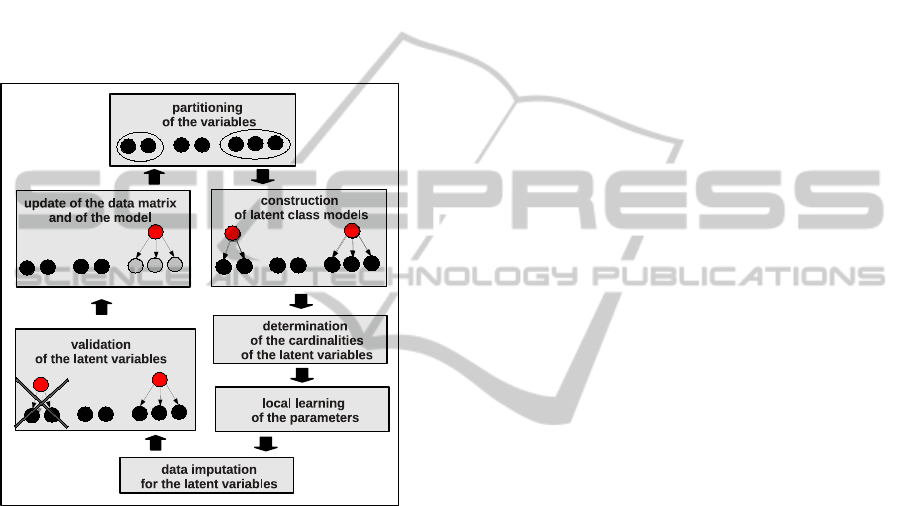
a scalable learning algorithm. Figure 2 depicts the
principle of this iterative algorithm, based on an as-
cending hierarchical clustering procedure.
Figure 2: Sketch of the iterative FLTM learning algorithm.
In the case of LD modeling, the observed variables
are the SNPs. The cardinality of these observed vari-
ables is equal to 3, which codes for minor homozy-
gosity, heterozygosity and major homozygosity. The
first iteration starts with the partitioning of the ob-
served variables into non overlapping clusters of pair-
wise highly dependent SNPs. No two variables are
allowed in the same cluster if their physical distance
on the genome is above a given threshold, δ. For each
cluster, an LCM is constructed whose child variables
are the variables in the cluster and whose latent vari-
able is created. The approach of (Mourad et al., 2011)
considers discrete latent variables whose cardinalities
may be different from one another; a heuristic is used
to determine the specific cardinality of each latent
variable. This specific cardinality is computed as an
affine function of the number of variables in the clus-
ter. Then, the parameters of each LCM are estimated
through the standard expectation-maximization pro-
cedure. Knowing the parameters of each LCM fur-
ther allows to impute the data corresponding to its la-
tent variable. Such imputed data are used by a val-
idation step; this step relies on a normalized mutual
information criterion to examine whether each novel
latent variable is sufficiently informative to subsume
its child variables. The data is updated with the val-
idated latent variables replacing their child variables.
The validated latent variables can thus be considered
as observed variables and an iteration begins anew.
This ascending process is iterated until no valid
cluster can be identified, or a single cluster of maxi-
mal size is obtained. Among other parameters of the
learning procedure, the clustering procedure plugged
in the algorithm is likely to impact the quality of the
LD modeling, and therefore the quality of the GWAS
performed downstream.
3.3 Performing a GWAS Guided by the
FLTM Model
Central to the use of the FLTM model for a GWAS
purpose are the request for data dimension reduction
and the motivation for a multilocus strategy. In the
study described here, we have implemented a multi-
locus GWAS strategy as follows: in the lowest lay-
ers, we traverse the forest top down, following a best-
first search strategy which only tests all child nodes
for the nodes whose association significances (i.e. p-
values) are below a threshold. The way to compute
this threshold is specific to the layer the variable be-
longs to. These child nodes are selected in turn with
respect to the appropriate threshold. The standard
Chi
2
test is applied to test a variable against the dis-
ease and to provide a pointwise p-value. We now
explain how to compute the thresholds for the vari-
ables in the lowest layers. To test statistical signifi-
cance, we consider a global threshold, say α = 5%.
The pointwise p-value of a variable is corrected based
on permutations applied on all the variables in this
variable’s latent layer. Thus, the correction is layer
specific. Statistical significance is assessed by testing
the condition (p-value
corrected
≤ α). The details and
justification for this correction will be provided in an
extended version of the article. On the other hand,
due to dimension reduction, the highest layers have a
low number of variables. No correction is applied for
these layers, from which we systematically select the
top most associated variables (e.g. β = 10%).
BIOINFORMATICS2015-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
8

4 THE CAST AND DBSCAN
PARTITIONING METHODS
Performing an optimal clustering is NP-hard (Ack-
erman and Ben-David, 2009). Therefore, heuristics
must be designed instead. In this paper, we focus
on two partitioning methods, CAST and DBSCAN,
to study how they impact LD modeling and a further
downstream GWAS analysis.
Since we address high-dimensionaldata, we could
not envisage the use of ascending hierarchical cluster-
ing, whose complexity scales in O(n
3
) where n is the
number of objects to be asssigned to clusters. More-
over, in contrast to AHC and k-means, another well
known partitioning method, CAST and DBSCAN do
not request the tuning of the number of clusters.
Partitioning objects into clusters relies on pairwise
distances (alternatively pairwise similarities). Stor-
ing a pairwise similarity matrix at the genome scale
is intractable. Thus, following (Mourad et al., 2011),
we acknowledge a physical constraint, δ, expressed in
kbp (kilobase pairs), in both implementations of the
CAST and DBSCAN methods. This constraint δ rep-
resents the physical distance on the genome beyond
which two objects (in our case two variables) are not
allowed in the same cluster. Additional calculus is re-
quired to estimate the distance between two variables
one of which at least is a latent variable.
The CAST (Cluster Affinity Search Technique) al-
gorithm was proposed in (Ben-Dor et al., 1999) and
is depicted in (Cahill, 2002). Its theoretical runtime
complexity scales in O(n
2
(log(n))
c
), with c some
constant, and its empirical complexity allows to han-
dle high-dimensional data. CAST is the native clus-
tering method used in the FLTM learning algorithm
depicted in (Mourad et al., 2011).
To decide cluster membership, CAST relies on
an affinity measure. In the implementation of CAST
adapted to FLTM learning, the binary similarity mea-
sure is assessed as the thresholded mutual informa-
tion (MI). A parameter q
pairwise
(e.g. 50%) allows
to compute the MI quantile (e.g. median) over the
pairs of variables whose physical distance is below
δ. This quantile allows to assign a binary similar-
ity (0/1), as in the native FLTM learning algorithm.
In this study, we also consider the unthresholded ver-
sion. These two CAST versions are denoted CAST
bin
and CAST
real
.
The DBSCAN (Density-Based Spatial Clustering
of Applications with Noise) algorithm was proposed
in (Ester et al., 1996). Its theoretical runtime com-
plexity is O(n
2
), where n is the number of objects
to be assigned to clusters. However, its empirical
complexity is known to be lower. To grow clusters,
DBSCAN relies on the merging of objects’ neighbor-
hoods. This method requires two parameters: R, the
maximum radius of the neighborhood to be consid-
ered, and N
min
, the minimum number of neighbors
needed for a cluster.
We have chosen DBSCAN as it is resistant to
noise. Besides, DBSCAN is known to be able to iden-
tify a cluster embedded in another cluster. On the
genome line, long-range LD corresponds to this sit-
uation.
5 METHODS
In this section, we first present the protocol used to
evaluate how much two partitions agree when fo-
cusing on the top most associated SNPs found by a
GWAS. Then, we motivate how we derived the so-
called reference partition (to be further defined). This
methodological section ends with the presentation of
the protocol used to compare the impact of the choice
of the partitioning method on the subsequent GWAS.
For this purpose, we rely on GWAS results published
in the literature.
5.1 Comparing Two Partitions
To cope with the genome scale, we were compelled to
select a simple method: we focused on the compari-
son of the partitions respectively obtained for the first
layer (SNPs) by two partitioning methods, and we ex-
amined how the top most associated SNPs identified
by a GWAS are distributed among the clusters.
The methods dedicated to the comparison of two
partitions may be categorized into three main groups
(Meila, 2005). Two groups attempt to map a partition
onto the other, either from set matching functions, or
from information theory-centered methods. The third
category relies on counting for how many pairs of el-
ements two partitions agree or disagree. The FLTM-
driven GWAS strategy is a multilocus strategy by def-
inition. In this multilocus GWAS framework, it is rel-
evant to analyze pair agreement between two parti-
tioning methods, for a selection of top most associ-
ated SNPs. A counting method fits well this purpose
of focusing on a subset of SNPs.
Given two partitions over the same set of objects,
and a pair of objects (in our case, a pair of variables),
an agreement means that the two partitions both group
the two variables in a cluster or both assign two differ-
ent clusters to the two variables. Given two partitions
P
1
and P
2
, let
• N
11
, the number of pairs both partitions assign to
one cluster,
ModelingGeneticalDatawithForestsofLatentTreesforApplicationsinAssociationGeneticsataLargeScale-Which
ClusteringMethodshouldBeChosen?
9

• N
00
, the number of pairs both partitions assign to
different clusters,
• N
10
, the number of pairs kept in the same cluster
by P
1
but splitted by P
2
,
• N
01
, the symmetric case of the latter.
From here, a large set of comparison measures is
available. We selected three measures to perform the
following comparisons: CAST
bin
versus CAST
real
,
CAST
bin
versus DBSCAN, CAST
real
versus DB-
SCAN, and each of the three methods CAST
bin
,
CAST
real
and DBSCAN versus the reference parti-
tion (to be defined in section 5.2). The comparison
measures selected are:
• the Rand index (Rand, 1971):
RI =
N
11
+ N
00
N
11
+ N
00
+ N
10
+ N
01
(1)
for which we used instead an adjusted corrected-
for-chance version (ARI =
RI−expected RI
maximum RI−expected RI
)
(for the detailed description, see (Hubert and Ara-
bie, 1985));
• the Mirkin distance (Mirkin, 1998):
MI =
S
P
1
+ S
P
2
− 2S
P
1
P
2
n
2
(2)
with
S
P
j
=
∑
cluster
i
∈P
j
| cluster
i
|
2
, j = 1, 2
S
P
1
P
2
=
∑
cluster
i
∈P
1
, cluster
j
∈P
2
| cluster
i
| | cluster
j
|
and n the number of objects to be assigned to clus-
ters and | S | the size of set S;
• the Fowlkes-Mallows index (Fowlkes and Mal-
lows, 1983):
FM =
r
N
11
N
11
+ N
10
·
N
11
N
11
+ N
01
. (3)
5.2 Deriving the Reference Partition
The reference partition intends to be the closest rep-
resentation of the underlying reality, that is the hap-
lotype blocks. We used the Haploview software pro-
gram (Gabriel et al., 2002) for this purpose. This ap-
plication allows to select commonly used block defi-
nitions to partition the genome into regions of strong
LD (Gabriel et al., 2002; Wang et al., 2002). As this
block generation is dedicated to handle genetical data,
Haploview can only be used for the first layer (ob-
served variables). This reason explains why the parti-
tioning method of the Haploview application has not
been plugged in the FLTM learning algorithm.
6 CROHN’S DISEASE GWAS
DATA
The Crohn’s disease data set we used is
made available by the WTCCC Consortium
(http://www.wtccc.org.uk/); it consists of 5009 indi-
viduals genotyped using the Affymetrix GeneChip
500K Mapping Array Set (3004 controls, 2005
cases). We performed the same data quality control
as the WTCCC. We excluded individuals, using
exactly the same criteria as the WTCCC ((WTCCC,
2007), page 26) (e.g. individuals with more than 3%
missing data across all SNPs; individuals sharing
more than 86% of identity with other ones). The rules
to exclude SNPs were also modelled after those of the
WTCCC (e.g. missing rate over 5%; if MAF (minor
allele frequency) under 5%, missing rate threshold
decreased to 1%) ((WTCCC, 2007), page 27).
In this paper, we focus on chromosome 2, known
to harbour SNPs with susceptibility towards Crohn’s
disease. The initial WTCCC data set describes 41400
SNPs. After the quality control step, our data con-
sisted of 38730 SNPs.
7 RESULTS AND DISCUSSION
The parameter t
CAST
(see details in (Cahill, 2002))
specific to the CAST method, whatever the version
(bin or real), was empirically set to 0.50. The pa-
rameter q
pairwise
specific to the CAST
bin
clustering
method was empirically chosen to be 50%. The N
min
and R parameters specific to DBSCAN were tuned to
2 and 0.2 respectively. The FLTM learning algorithm
requires the setting of six parameters. We systemati-
cally evaluated the coefficients of the affine function
used to determine the cardinality of each latent vari-
able, ℓ
1
and ℓ
2
, in [0.2, 0.3, 0.4, 0.5] × [1, 2]. We ob-
served no differences between the eight settings, with
regard to the sizes and contents of the clusters. Thus,
ℓ
1
and ℓ
2
were set 0.5 and 1. Following (Mourad
et al., 2011), we fixed the maximum cardinality as
20, the physical distance constraint δ as 45 kbp and
the number of restarts of the stochastic expectation-
maximization procedure as 10. The threshold for the
quality control of the candidate latent variables was
set to a low value, 0.01. The GWAS thresholds α and
β were fixed to 5% and 10%. The study was con-
ducted using a 3.3 GHz processor. We had to adapt
the generic versions of the CAST and DBSCAN al-
gorithms, to store a sparse similarity matrix instead
of a pairwise similarity matrix (see section 4).
BIOINFORMATICS2015-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
10
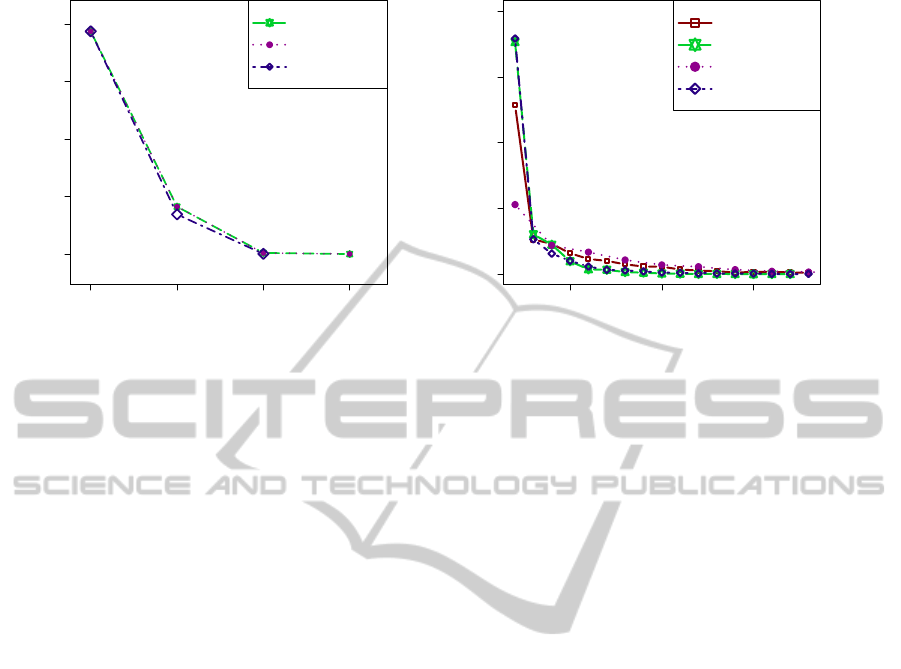
Layer
38730
8241
190
2
8243
178
2
6890
24
0 10000 20000 30000 40000
0 1 2 3
CAST
real
CAST
bin
DBSCAN
Number of variables
5 10 15
0.0 0.2 0.4 0.6 0.8
Number of variables
Density
Haploview
CAST
real
CAST
bin
DBSCAN
(a) (b)
Figure 3: Impact of the choice of the partitioning methods CAST
bin
, CAST
real
and DBSCAN on the structure of the FLTM
model. (a) Impact on the number of variables per layer. (b) Impact on the sizes of the clusters for the first layer (observed
layer).
7.1 FLTM Architectures
On average, the running time observed for FLTM
learning with each clustering method is in the order of
60 hours. A closer examination shows that clustering
and other operations only required at most 1 minute
for each layer, and that practically all the running time
was spent in the expectation-maximization procedure
(see section 3.2). Moreover, it is likely that the pres-
ence of a few clusters of large size (size up to 50)
severely increases running times for the expectation-
maximization procedure.
We first analyze the impact of the partitioning
method on the structure of the FLTM model con-
structed prior to a GWAS. Figure 3 (a) compares
the impacts of the three partitioning methods on the
data dimension reduction. We observe that for any
layer, the total number of latent variables created us-
ing CAST
real
is always greater than that created using
DBSCAN. Moreover, layer 3 does not exist for DB-
SCAN whereas it exists for CAST
bin
and CAST
real
.
Indeed, for DBSCAN, no more variables can be
grouped in layer 2: all candidate clusters are sin-
gletons. The numbers of variables in layers 1 and 3
are either very close or similar between CAST
bin
and
CAST
real
. Again, among the three methods, the num-
bers of variables in layer 2 are the closest for CAST
bin
and CAST
real
.
Figure 3 (b) provides the histogram for the sizes
of the clusters in the first (observed) layer, for each
of the three partitioning methods, together with the
histogram of the reference Haploview partitioning. It
has to be mentioned that, for reasons of presentation,
the histograms have been truncated. Very few clus-
ters of large sizes are observed: the maximum sizes
observed are 18, 45 and 50 for CAST
real
, DBSCAN
and CAST
bin
, respectively. Such clusters would nor-
mally appear far apart on the right section of Figure 3
(b).
First, we observe that from size 3, the CAST
bin
curve is slightly above the CAST
real
and DBSCAN
curves. Besides, the latter curves are nearly super-
imposed. Finally, we note that from size 3, the curve
relative to the reference partitioning is located slightly
below that of CAST
bin
, on the one hand, and slightly
abovethe quasi superimposed curves of CAST
real
and
DBSCAN, on the other hand.
Therefore, the general conclusion to draw for
this section is the propensity for DBSCAN to pro-
duce a lower number of variables than CAST
bin
and
CAST
real
, but with no clear impact on the differences
between the cluster size histograms.
7.2 Comparison of the Partitioning
Methods in a GWAS Context
In a GWAS context, we wish to focus in pri-
ority on pairs of SNPs selected among the top
SNPs found most associated with the studied dis-
ease. The standard tool PLINK was used to
identify these top SNPs (Purcell et al., 2007)
(http://pngu.mgh.harvard.edu/purcell/plink/). Rely-
ing on PLINK, we performed a single-SNP GWAS on
the WTCCC data set relative to chromosome 2. The
association test used was the Chi
2
. We have extended
the agreement analysis of two partitions to embedded
ModelingGeneticalDatawithForestsofLatentTreesforApplicationsinAssociationGeneticsataLargeScale-Which
ClusteringMethodshouldBeChosen?
11
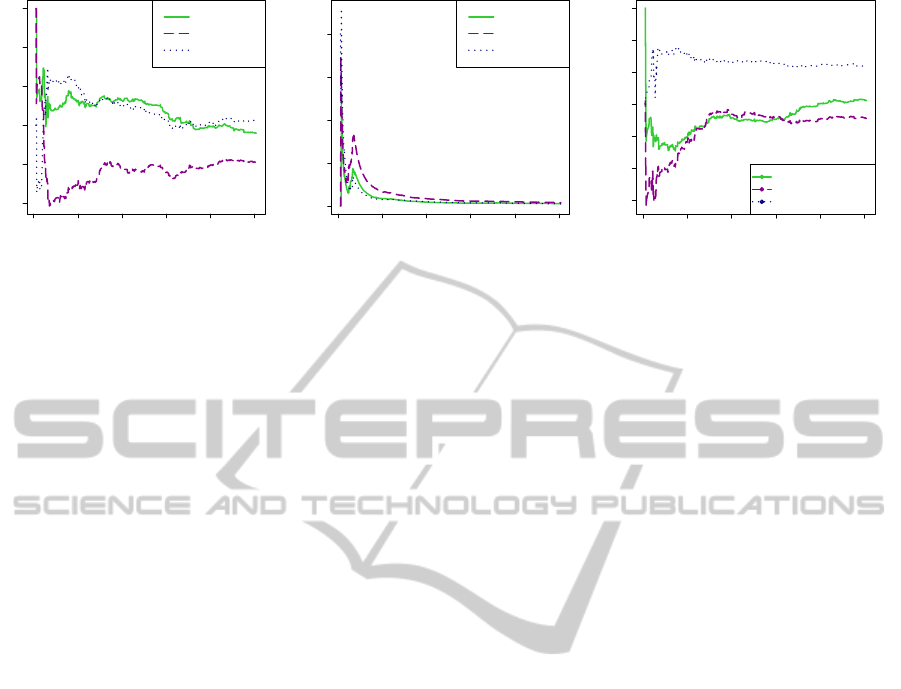
0 200 400 600 800 1000
0.5 0.6 0.7 0.8 0.9 1.0
Top N Variables
CAST
real
CAST
bin
DBSCAN
Adjusted Rand Index
0 200 400 600 800 1000
0.00 0.02 0.04 0.06 0.08
Top N Variables
CAST
real
CAST
bin
DBSCAN
Mirkin Distance
0 200 400 600 800 1000
0.4 0.5 0.6 0.7 0.8 0.9 1.0
Top N Variables
CAST
real
− CAST
bin
DBSCAN−CAST
bin
DBSCAN−CAST
real
Adjusted Rand Index
(a) (b) (c)
Figure 4: Agreement of two partitioning methods, in a GWAS context. (a) and (b) Agreement of a partitioning method with
the reference block partitioning method used by Haploview. Comparison for the partitioning methods CAST
bin
, CAST
real
and DBSCAN. Impact of the number of top SNPs considered on the agreement. The top SNPs considered are those found
most significantly associated by a standard single-SNP GWAS. (a) Adjusted Rand index. (b) Mirkin distance. (c) Pairwise
comparison of the partitioning methods CAST
bin
, CAST
real
and DBSCAN. Impact of the number of top SNPs considered on
the agreement. Adjusted Rand index.
sets of associated SNPs, increasing the size of the set
of top associated SNPs up to 1000.
Figures 4 (a) and (b) compare the partioning meth-
ods CAST
bin
, CAST
real
and DBSCAN together with
Haploview, following two of the three comparison cri-
teria described in section 5.1.
The adjusted Rand index is all the higher as the
agreement between two partitioning methods is high.
Thus, we observe that CAST
bin
does not agree with
the reference (Haploview) partitioning as well as
CAST
real
and DBSCAN. This specificity of CAST
bin
is explained by the conversionof real mutual informa-
tion values into binary values (see the role of param-
eter q
pairwise
in section 4). This discretization there-
fore entails slightly larger cluster sizes for CAST
bin
,
as seen in section 7.1.
On the left section of Figure 4 (a), the index
is computed from few top SNPs. We observe that
CAST
bin
and CAST
real
show a high Rand index in
contrast to DBSCAN. However, in a GWAS context,
we do not wish to examine only, say, the 20 top sig-
nificantly associated SNPs. Thus, the most relevant
section to focus on is around 50-100 top SNPs. In
this latter section of Figure 4 (a), we observe that
the CAST
real
and DBSCAN curves are comparatively
close and clearly located higher than the CAST
bin
curve. This trend is observed up to the 1000 top most
associated SNPs.
In Figure 4 (b), a low Mirkin distance indicates
a high agreement between two partitioning meth-
ods. The observations in Figure 4 (b) confirm that
CAST
bin
’s agreement with Haploviewclustering is al-
ways worse than the other two methods’. We have
not shown the results for the Fowlkes-Mallows index
as the curves obtained are quasi superimposable with
those plotted for the adjusted Rand index.
The first general conclusion to draw from this first
series of agreement comparisons on the Crohn’s dis-
ease data set is that DBSCAN and CAST
real
show
a high level agreement with Haploview partitioning,
both being quite clearly better than CAST
bin
.
Figure 4 (c) displays the results for pairwise com-
parisons: CAST
real
versus CAST
bin
, DBSCAN ver-
sus CAST
bin
and DBSCAN versus CAST
real
. Ac-
cording to the adjusted Rand index, DBSCAN and
CAST
real
show a high agreement. Given our pre-
vious observations, we expected that CAST
bin
and
CAST
real
would show a low level agreement, which
is confirmed. DBSCAN and CAST
bin
yield partitions
that almost always disagree more than for the two for-
mer couples of partitioning methods. This trend is
confirmed with the Mirkin distance and the Fowlkes-
Mallows index (results not shown).
As a second general conclusion of this section,
we cross-confirm one of our previous observations:
DBSCAN and CAST
real
each show a high agree-
ment with Haploview. This fact is therefore also
reflected by a high agreement between DBSCAN and
CAST
real
.
7.3 FLTM-driven GWASs
In Figure 5, the comparison of plots (a) to (c) and
plot (d) shows how the dimension reduction allows
to pinpoint the potentially most interesting regions
on the genome. Thus, ”sparse” association profiles
are produced, as opposed to the dense output of the
standard single-SNP GWAS.
The two putative causal SNPs located on chro-
mosome 2 respectively reported in the WTCCC
study (WTCCC, 2007) and in (Barrett et al., 2008)
BIOINFORMATICS2015-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
12
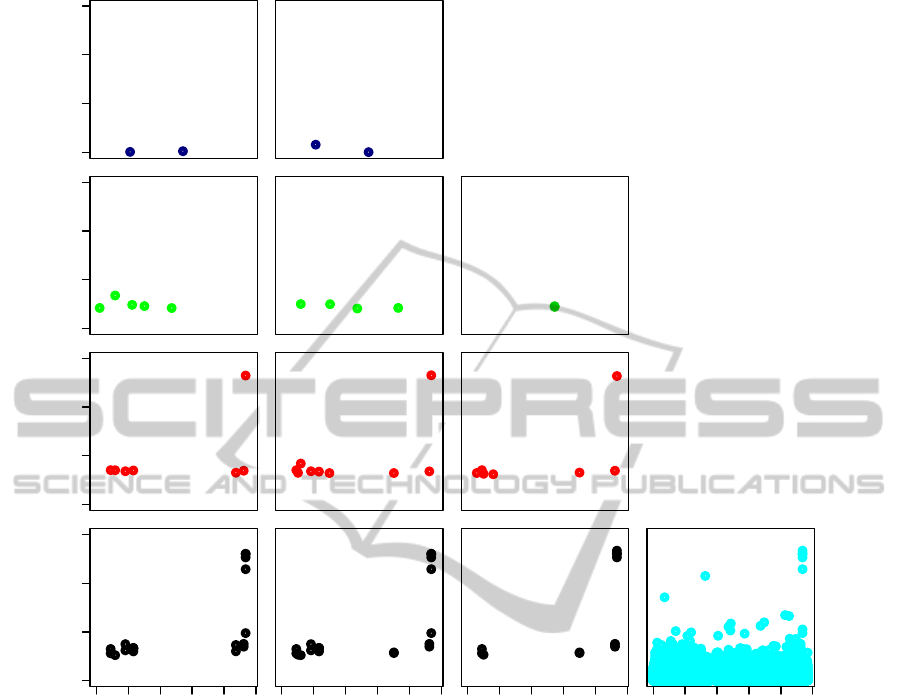
0 5 10 15
(a) CAST
bin
−log
10
(p−value)
Layer 3
0 5 10 15
−log
10
(p−value)
Layer 2
0 5 10 15
−log
10
(p−value)
Layer 1
0 50 100 200
0 5 10 15
−log
10
(p−value)
Layer 0
Mbp
(b) CAST
real
Layer 3
Layer 2
Layer 1
Layer 0
0 50 100 200
Mbp
(c) DBSCAN
Layer 2
Layer 1
Layer 0
0 50 100 200
Mbp
0 50 100 200
(d) Single−SNP GWAS
Mbp
Figure 5: Impact of the choice of the partitioning method on the multilocus GWAS results. For the FLTM-based GWASs ((a)
to (c)), one ”sparse” association profile is displayed for each layer, as not all variables in a layer are examined. The single-SNP
GWAS in (d) was performed using the gold standard PLINK (Purcell et al., 2007). Its output only deals with variables in layer
0 (observed variables). All plots show initial (i.e. non corrected) p-values.
are identified by the three FLTM-driven GWASs.
Given that we used the same data set as in (WTCCC,
2007), one of the two results was expected. However,
this result was not guaranteed, because of the data
dimension reduction and of the subsumption involved
in an FLTM-driven GWAS. Besides, it must be
highlighted that the study in (Barrett et al., 2008)
analyzed 8059 individuals (3230 cases and 4829
controls), whereas the WTCCC data set describes a
population of size 5009. Table 1 shows that CAST
bin
and CAST
real
capture exactly the same four highly
associated SNPs through the latent variables L
1
and
L
2
, belonging to layer 1. These variables are the
right-most latent variables in layer 1, on the plots (a)
and (b) of Figure 5. The virtual location of a latent
variable is computed as the average of the locations of
its child variables. Thus, the location of L
1
(or L
2
) is
233837691 bp. The p-values computed for L
1
and L
2
differ since the data imputed for these latent variables
differ. For either CAST
bin
or CAST
real
, the SNP
published in (WTCCC, 2007) is not grouped with
other SNPs into a cluster, in contrast to DBSCAN.
Table 1 shows that for DBSCAN, the latent variable
L
3
subsumes SNPs among which are the two already
published putative causal SNPs. L
1
and L
3
share
three highly associated SNPs, including the putative
causal SNP published in (Barrett et al., 2008). The
virtual location of L
3
is 233830355 bp. We can see
that L
1
captures LD on a slightly wider range than
L
3
, since the regions encompassed by the former and
the latter variables spread over 29292 and 21571 bp,
respectively.
A more thorough analysis of the Affymetrix ar-
ModelingGeneticalDatawithForestsofLatentTreesforApplicationsinAssociationGeneticsataLargeScale-Which
ClusteringMethodshouldBeChosen?
13

Table 1: Analysis of the latent variables in layer 1 found significantly associated with Crohn’s disease, by the three FLTM-
driven GWASs with plug-in CAST
bin
, CAST
real
and DBSCAN, respectively. For each clustering method, the latent variable
is described on the first line. On the following lines, the highly associated SNPs subsumed by this latent variable are depicted.
The identifier of each SNP is provided (rsXXXXXXX). The • character highlights the SNPs which are common children of
latent variables L
1
(or L
2
) and L
3
. ∗ Note that the association tests used may differ between studies.
Clustering method Variable Location p-value p-value reported in another study
∗
CAST
bin
latent L
1
233837691 (1) 5.86× 10
−14
rs6752107 233826187 • (2) 9.65× 10
−14
(3)
rs6431654 233826508 • (2) 9.96× 10
−14
(4)
rs3828309 233845149 • (2) 2.30× 10
−13
(5) 2× 10
−32
(Barrett et al., 2008)
rs3792106 233855479 (2) 3.70× 10
−12
CAST
real
latent L
2
see (1) 5.52 × 10
−14
see (2)
DBSCAN
latent L
3
233830355 6.58× 10
−14
rs10210302 233823578 4.60 × 10
−14
7× 10
−14
(WTCCC, 2007)
rs6752107 233826187 • see (3)
rs6431654 233826508 • see (4)
rs3828309 233845149 • see (5) 2× 10
−32
(Barrett et al., 2008)
ray indicates that the region encompassed by L
1
,
[233826187, 233855479], contains four highly as-
sociated SNPs, interspersed with three non associ-
ated SNPs. Similarly, the interval covered by L
3
,
[233823578, 233845149], contains eight SNPs, in-
cluding four non associated SNPs. Clearly, among
the four highly associated SNPs pinpointed by each
of L
1
and L
3
, respectively three and two SNPs are
highly associated with the disease because they are
in LD with a putative causal SNP (see Table 1). How-
ever, not every SNP close to a putative causal SNP
has been incorporated in the cluster subsumed by L
1
,
L
2
or L
3
. To confirm the relevance of the clustering
performed, an in-depth examination shows that these
former close SNPs that are not in LD with putative
causal SNPs are found poorly associated with the dis-
ease (in the order of 10
−1
). Importantly,even the SNP
flanking on the left the causal putative SNP published
in (Barrett et al., 2008) and having a p-value equal to
1.32 × 10
−5
, was not retained in L
1
or L
3
’s cluster.
This observation shows that a fine-grain clustering is
achieved for each of the three partitioning methods.
Therefore, a first remarkable result is that the sub-
sumption process does not hinder the informative-
ness of L
1
, L
2
and L
3
: L
1
, L
2
and L
3
are still found
highly associated with the disease (5,86 × 10
−14
,
5,52 × 10
−14
, 6.58× 10
−14
respectively).
Moreover, a second remarkable result is ob-
tained. The standard GWAS (Figure 5 (d)) iden-
tifies two SNPs with a high statistical signifi-
cance (rs13394205, located at around 18 Mbp
(17849508), and rs11887827, located at around 81
Mbp (81519665)). The p-values of these two
SNPs are respectively 2.28× 10
−9
and 1.81× 10
−11
.
None of these SNPs were reported in former stud-
ies (WTCCC, 2007) and (Barrett et al., 2008), which
identified them as false positives. In the layers 0 of
the plots (a) to (c) of Figure 5, none of these two
SNPs either appears. The reason lies in that during the
top down traversal of the FLTM, the parents of these
SNPs are detected as not significantly associated with
the studied disease. Consequently, the descendants of
these latent variables are not examined (and not dis-
played in the sparse outputs). Therefore, the FLTM-
driven GWAS strategy exerts an efficient control of
the number of false positives. Furthermore, all layers
potentially allow to exert such a control, with a prun-
ing effect on the forest structure guiding the GWAS.
In the context of this study, the general conclu-
sion to draw from this section is that the three FLTM-
driven GWASs capture the SNPs reported associated
by two other studies and correctly detect false positive
associations. Second, the differences reported in sec-
tions 7.1 and 7.2 between CAST
bin
and the two other
clustering methods do not impact the quality of the
corresponding FLTM-driven GWAS.
8 CONCLUSION AND
PERSPECTIVES
In this paper, we have studied the impact of the choice
of the clustering method to be plugged in the FLTM
learning algorithm, for the purpose of a GWAS ap-
plication. We have started analyzing this impact fo-
cusing on two scalable clustering methods, adding a
relaxed variant of one of them. For this purpose, a
methodological framework has been designed, which
allows to compare the three clustering methods ac-
cording to the following viewpoints: (1) effective
ability to split or group the top associated SNPs,
according to the underlying linkage disequilibrium
structure; (2) data dimension reduction and associated
risk of missing significant results through subsump-
BIOINFORMATICS2015-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
14

tion; (3) relevance of the partitioning method to guide
an FLTM-based GWAS pinpointing regions with sig-
nificantly associated SNPs. The CAST
bin
clustering
method was shown slightly different from CAST
real
and DBSCAN, from the clustering viewpoint. How-
ever, this difference was not reflected by a difference
in GWASs’ performances. Therefore, to the initial
question ”Which clustering method should be cho-
sen”, the answer for the Crohn’s disease WTCCC data
set relative to chromosome 2 would rather prioritize
easiness in tuning parameters. In our experiments so
far, the FLTM learning algorithm seems robust to the
choice of the clustering method, provided that the in-
trinsic parameters of the latter are appropriately set.
Further works include extending the current analysis
to other chromosomes, for the WTCCC data set, as
well as to other diseases, and extending our analysis
to other clustering methods.
It was the first time that the FLTM learning algo-
rithm was run on real GWAS data. It is questionable
whether the present study should be complemented by
intensive experiments run on simulated GWAS data
sets. Given the high processing times required as soon
as GWASs are addressed, and the recurring question
of generating sufficiently realistic GWAS data, a less
systematic approach, encompassing more diseases,
seems wholly relevant.
Finally, to return to the multilocus aspect of the
type of GWAS addressed here, one of our next tasks
is to compare the FLTM-based GWAS strategy with
the few other scalable multilocus approaches existing,
including BEAGLE (Browning and Browning, 2007).
ACKNOWLEDGEMENTS
The project SAMOGWAS (Specific Advanced MOd-
els for Genome Wide Association Studies) is sup-
ported by the French National Research Agency
(Agence Nationale de la Recherche, ANR). The au-
thors are also grateful to the Wellcome Trust Case
Control Consortium for providing the GWAS data
used in this study.
REFERENCES
Abel, H. and Thomas, A. (2011). Accuracy and Com-
putational Efficiency of a Graphical Modeling Ap-
proach to Linkage Disequilibrium Estimation. Statis-
tical Applications in Genetics and Molecular Biology,
10(1):Article 5.
Ackerman, M. and Ben-David, S. (2009). Clusterability: a
Theoretical Study. In Dyk, D. and Welling, M., ed-
itors, Twelfth International Conference on Artificial
Intelligence and Statistics (AISTATS09), Journal of
Machine Learning Research, Proceedings Track, vol-
ume 5, pages 1–8.
Balding, D. (2006). A Tutorial on Statistical Methods for
Population Association Studies. Nature Reviews Ge-
netics, 7(10):781–791.
Barrett, J., Hansoul, S., Nicolae, et al. (2008). Genome-
wide Association Defines more than 30 Distinct Sus-
ceptibility Loci for Crohn’s Disease. Nature Genetics,
40(8):955–962.
Ben-Dor, A., Shamir, R., and Yakhini, Z. (1999). Clus-
tering Gene Expression Patterns. In Third Annual In-
ternational Conference on Research in Computational
Molecular Biology (RECOMB99), pages 33–42.
Browning, B. and Browning, S. (2007). Efficient Multilo-
cus Association Testing for Whole Genome Associ-
ation Studies Using Localized Haplotype Clustering.
Genetic Epidemiology, 31:365–375.
Cahill, J. (2002). Error-Tolerant Clustering of Gene Mi-
croarray Data. Bachelors Honors Thesis, Boston Col-
lege, Massachusetts.
Ester, M., Kriegel, H.-P., Sander, J., and Xu, X. (1996). A
Density-Based Algorithm for Discovering Clusters in
Large Spatial Databases with Noise. In Second In-
ternational Conference on Knowledge Discovery and
Data Mining (KDD96), pages 226–231.
Fowlkes, E. and Mallows, C. (1983). A Method for Com-
paring Two Hierarchical Clusterings. Journal of the
American Statistical Association, 78(383):553–569.
Gabriel, S., Schaffner, S., Moore, J., et al. (2002). The
Structure of Haplotype Blocks in the Human Genome.
Science, 296(5576):2225–2229.
Gibbs, R., Belmont, J., Hardenbol, P., et al. (2003). The In-
ternational HapMap Project. Nature, 426(6968):789–
796.
Hubert, L. and Arabie, P. (1985). Comparing Partitions.
Journal of Classification, 2(1):193–218.
Meila, M. (2005). Comparing Clusterings: an Axiomatic
View. In Twenty-second International Conference on
Machine Learning (CML05), ACM, pages 577–584.
Mirkin, B. (1998). Mathematical Classification and Clus-
tering: from How to What and Why. Classification,
Data Analysis, and Data Highways, 690:172–181.
Mourad, R., Sinoquet, C., and Leray, P. (2011). A Hierar-
chical Bayesian Network Approach for Linkage Dise-
quilibrium Modeling and Data-dimensionality Reduc-
tion prior to Genome-wide Association Studies. BMC
Bioinformatics, 12:16+.
Pritchard, J. and Przeworski, M. (2001). Linkage Disequi-
librium in Humans: Models and Data. The American
Journal of Human Genetics, 69(1):1–14.
Purcell, S., Neale, B., Todd-Brown, K., et al. (2007).
PLINK: a Toolset for Whole-genome Association and
Population-based Linkage Analysis. The American
Journal of Human Genetics, 81(3):559–575.
Rand, W. (1971). Objective Criteria for the Evaluation of
Clustering Methods. Journal of the American Statisti-
cal Association, 66(336):846–850.
ModelingGeneticalDatawithForestsofLatentTreesforApplicationsinAssociationGeneticsataLargeScale-Which
ClusteringMethodshouldBeChosen?
15

The 1000 Genomes Project Consortium (2010). A Map of
Human Genome Variation from Population-scale Se-
quencing. Nature, 467(7319):1061–1073.
Verzilli, C., Stallard, N., and Whittaker, J. (2006).
Bayesian Graphical Models for Genome-wide Asso-
ciation Studies. The American Journal of Human Ge-
netics, 79:100–112.
Wang, N., Akey, J., Zhang, K., Chakraborty, R., and Jin,
L. (2002). Distribution of Recombination Crossovers
and the Origin of Haplotype Blocks: the Interplay
of Population History, Recombination, and Muta-
tion. The American Journal of Human Genetics,
71(5):1227–1234.
WTCCC (2007). Wellcome Trust Case Control Con-
sortium. Genome-wide Association Study of 14,000
Cases of Seven Common Diseases and 3,000 Shared
Controls. Nature, 447(7145):661–678.
BIOINFORMATICS2015-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
16
