
PERFECTOS-APE
Predicting Regulatory Functional Effect of SNPs by Approximate P-value
Estimation
Ilya E. Vorontsov
1
, Ivan V. Kulakovskiy
1,2
, Grigory Khimulya
1
, Daria D. Nikolaeva
3
and Vsevolod J. Makeev
1,2,4
1
Department of Computational Systems Biology, Vavilov Institute of General Genetics, Gubkina str. 3, Moscow, Russia
2
Laboratory of Bioinformatics and Systems Biology, Engelhardt Institute of Molecular Biology,
Vavilova str. 32, Moscow, Russia
3
Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, Moscow, Russia
4
Department of Medical and Biological Physics, Moscow Institute of Physics and Technology, Moscow Region, Russia
Keywords:
Single Nucleotide Polymorphism, SNP, Single Nucleotide Variant, SNV, P-value, Transcription Factor
Binding Site, TFBS, Position Weight Matrix, PWM, PSSM, Transcriptional Regulation.
Abstract:
Single nucleotide polymorphisms (SNPs) and variants (SNVs) are often found in regulatory regions of hu-
man genome. Nucleotide substitutions in promoter and enhancer regions may affect transcription factor (TF)
binding and alter gene expression regulation. Nowadays binding patterns are known for hundreds of human
TFs. Thus one can assess possible functional effects of allele variations or mutations in TF binding sites using
sequence analysis.
We present PERFECTOS-APE, the software to PrEdict Regulatory Functional Effect of SNPs by Approxi-
mate P-value Estimation. Using a predefined collection of position weight matrices (PWMs) representing TF
binding patterns, PERFECTOS-APE identifies transcription factors whose binding sites can be significantly
affected by given nucleotide substitutions. PERFECTOS-APE supports both classic PWMs under the position
independency assumption, and dinucleotide PWMs accounting for the dinucleotide composition and correla-
tions between nucleotides in adjacent positions within binding sites.
PERFECTOS-APE uses dynamic programming to calculate PWM score distribution and convert the scores
to P-values with an optional binary search mode using a precomputed P-value list to speed-up the com-
putations. Software is written in Java and is freely available as standalone program and online tool:
http://opera.autosome.ru/perfectosape/.
We have tested our algorithm on several disease associated SNVs as well as on a set of cancer somatic muta-
tions occurring in intronic regions of the human genome.
1 INTRODUCTION
Single nucleotide variants (SNVs) are the most stud-
ied variations of the human genome. Modern
genome-wide association studies link different SNV
alleles with different phenotypes, including disease
susceptibility. High-throughput technologies become
cheaper and push forward sequencing of personal
genomes and detection of individual genome variants.
However, proper interpretation of the sequencing data
remains a challenge. Most of the detected SNVs are
located outside of protein coding regions including a
special class of SNVs found in gene regulatory re-
gions. Among those an important subclass is formed
by promoter and enhancer SNVs, which do not al-
ter protein sequence or structure but possibly affect
gene expression. Such SNVs may affect transcrip-
tion through alterations in transcription factor binding
sites.
During the past 10 years a number of tools (Mac-
intyre et al., 2010; Manke et al., 2010; Barenboim
and Manke, 2013; Riva, 2012; Teng et al., 2012; An-
dersen et al., 2008; Khurana et al., 2013) were devel-
oped for computational analysis of regulatory SNVs.
Basic algorithms were simply predicting TFBS over-
lapping an SNV position (Ponomarenko et al., 2001).
More sophisticated tools compare predicted affinitites
of TFs binding to binding sites for different homolo-
102
E. Vorontsov I., V. Kulakovskiy I., Khimulya G., D. Nikolaeva D. and J. Makeev V..
PERFECTOS-APE - Predicting Regulatory Functional Effect of SNPs by Approximate P-value Estimation.
DOI: 10.5220/0005189301020108
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2015), pages 102-108
ISBN: 978-989-758-070-3
Copyright
c
2015 SCITEPRESS (Science and Technology Publications, Lda.)

gous alleles (Manke et al., 2010) and estimate statis-
tical significance of the difference (Macintyre et al.,
2010). All existing tools use the well-studied but ba-
sic model of transcription factor binding sites, the po-
sition weight matrix. Nowadays high-throughput data
such as ChIP-Seq allows producing advanced mod-
els (Kulakovskiy et al., 2013a; Mathelier and Wasser-
man, 2013), since the volume of data makes it pos-
sible to train models with more parameters without
the risk of overfitting (Levitsky et al., 2014; Mathe-
lier and Wasserman, 2013). Here we present a novel
software, PERFECTOS-APE, to predict how differ-
ent alleles of SNVs or SNPs may alter affinity of tran-
scription factor binding sites modelled by basic and
advanced approaches.
2 METHODS
The core idea of our method is similar to that of (Mac-
intyre et al., 2010; Manke et al., 2010): the algorithm
estimates the statistical significance (the P-value) of
predicted TF binding sites overlapping an SNV. Then
it checks if TFBS binding P-values calculated for dif-
ferent homologous alleles differ enough. Extremely
small or large ratios of P-values indicate the cases
where the binding site exists only for one of the two
alleles.
2.1 TFBS Models and P-value
Estimation
We use basic and dinucleotide position weight matri-
ces (PWMs) as TFBS models. PWM quantitatively
describes which nucleotides are preferred at which
position. Classic PWM M is a matrix 4 ×k which
defines a score function on k-mers α
1
...α
k
upon nu-
cleotide alphabet V = {A,C, G, T }:
score(M, α
1
...α
k
) =
k
∑
i=1
M(α
i
,i) (1)
Similarly to PWM, the dinucleotide position
weight matrix (diPWM) D is a matrix 16 ×(k −1)
where each element provides score for a pair of con-
sequent nucleotides, where adjacent nucleotide pairs
overlap:
score(D,α
1
...α
k
) =
∑
i=1...k−1
D(α
i
α
i+1
,i) (2)
Dinucleotide PWM (diPWM) takes into account
dependent contributions of adjacent nucleotides.
Scoring function with a score threshold defines
the set of recognized words scoring no less than the
threshold. Higher scores correspond to better TF-
DNA recognition, but it’s not trivial to define a uni-
fied scale for different PWMs. To this end a P-value
of a given word for a given PWM is defined as the
probability that a random word scores not less than
the given threshold. In other words, the P-value cor-
responds to the area under the right tail of the PWM
score distribution. For example, if a random model
generates any word with equal probability, than the
P-value is the normalized cumulative count of words
with scores passing the threshold.
Let’s assume the words are generated by an i.i.d.
random model with p
α
frequencies of individual nu-
cleotides for a PWM model or by a Markov(1) model
for a diPWM.
Given a PWM score threshold t, P-value for this
PWM (denoted M) is:
pvalue =
∑
w∈V
k
score(M,w)≥t
P(w) (3)
Here P(w) is the probability of a word w to be
generated by the background model (either i.i.d. or
Markov(1)).
To convert PWM scores to P-values we uti-
lize a simplified dynamic programming approach
(ScoreDistribution algorithm) originally presented by
H.Touzet and J.Varr
´
e (Touzet et al., 2007).
The idea is to discretize PWM elements and pro-
duce overall score distribution in a form of a hash (un-
like the original approach here we use a predefined
discretization level). This allows finding approximate
P-value for a given score in reasonable time.
Each key in the hash is a score and the correspond-
ing stored value is a probability to obtain this score.
This hash can be constructed through recalculation of
the score distribution gradually increasing the length
of words:
H
0
(S) = [S = 0] ·1 (4)
H
l+1
(S
0
) =
∑
α∈V
∑
S:M(α,l+1)+S=S
0
H
l
(S)p
α
(5)
For a dinucleotide position weight matrix
(diPWM) the score distribution can be obtained
by a similar method. For a dinucleotide model D
(score, last-letter) pairs can be used as keys of the
hash:
H
1
(S,α) = [S = 0] ·
∑
γ∈V
p
γα
(6)
H
l+1
(S
0
,α
l+1
) =
∑
α
l
∈V
∑
S:D(α
l
α
l+1
,l)+S=S
0
H
l
(S,α
l
)p
α
l+1
|α
l
(7)
Here p
αβ
is the background probability of the din-
ucleotide αβ and p
β|α
is the background probability
PERFECTOS-APE-PredictingRegulatoryFunctionalEffectofSNPsbyApproximateP-valueEstimation
103

of the nucleotide β under condition that the previous
letter was α:
p
β|α
=
p
αβ
∑
γ
p
αγ
(8)
At the last step score distributions of words ending
with different nucleotides are summed to obtain the
final score distribution:
H
k
(S) =
∑
α∈V
H
k
(S,α) (9)
2.2 Speeding-up P-value Estimation
In practice it is convenient to analyze multiple SNVs.
Hundreds of thousands or even millions candidate
variants can be subjected to analysis thanks to the per-
sonal genomics data.
In this case P-value estimation can be accelerated
using precomputed score distributions for each TFBS
model in a given collection. The PWM score dis-
tribution is discrete; thus, it’s possible to store the
P-value for each achievable PWM score. The P-value
is a monotonically decreasing function depending on
scores. Thus the list of (score, P-value) pairs can be
sorted with respect to both the P-value and the score.
Given the word score t, binary search can be uti-
lized to find a pair of consequent scores t
1
,t
2
such that
t
1
≤t ≤ t
2
and thus the P-value p associated with the
score t lies in range p
1
≥ p ≥ p
2
.
A value between p
1
and p
2
can be taken as an es-
timate of the true P-value; we use the geometric mean
p ≈
√
p
1
p
2
.
Since in practice we don’t need a precise P-value,
but a reasonable approximation, we select admissible
P-value relative error δ to further improve computa-
tion time. From a given score distribution we take
not the whole list of (score, P-value) pairs but only
a subset such that P-values differ in (1 + δ) times:
p
0
, p
0
/(1 + δ), p
0
/(1 + δ)
2
,... for any p
0
. With the
help of this list we can estimate P-value with a given
relative error δ. We use the fixed discretization level
for PWMs at the distribution estimation step. We call
the whole procedure Approximate P-value Estimation
(APE).
To measure possible functional effect of allele
variants we compute fold change of P-values corre-
sponding to the binding sites with these allele vari-
ants (see the next section for details). In turn, the fold
change relative error will be not greater than
1+δ
1−δ
.
2.3 Predicting Functional Effect of
SNVs
To predict possible functional effect of a given SNV,
PERFECTOS-APE (1) scans the region overlapping
SNV position with (di)PWM model predicting bind-
ing sites for given nucleotide variants; (2) selects the
best (di)PWM prediction for each allele; (3) computes
P-values for the binding sites detected for each al-
lele variant. If any of P-values is small enough (i.e.
there is an allele corresponding to the putative bind-
ing site) and P-values differ significantly (i.e. the ratio
passes a fold change threshold t
FC
) we state that the
SNV may play a regulatory role through disruption or
emergence of the transcription factor binding site.
3 ALGORITHM COMPLEXITY
Given a motif of length k, a scanned region around
SNV should contain no less than 2k −1 nucleotides
(the SNV position with the flanking genomic re-
gions). The first stage of the algorithm calculates
scores for each site position in both strand orienta-
tions relative to the given DNA sequence. It takes
O(k
2
) operations to compute the scores (2k score esti-
mations with each score estimation of O(k) complex-
ity).
The second stage computes P-values either by cal-
culating score distribution using dynamic program-
ming or via binary search in a precomputed list.
Dynamic programming involves k steps. At each
step the algorithm updates the hash H of the score dis-
tribution (score to probability mapping). For a PWM
each update has O(|H||V |) complexity. For a diPWM
each update consists of O(|H||V |
2
) operations and
k −1 updates are to be done. Here |H| is a number of
elements in the hash and |V | is an alphabet size which
is equal to 4. Hash size can be roughly estimated as
|H| ≤ (max score −min score) · discretization rate
for a motif model discretized as:
M
discretized
(α,i) =
d
M(α,i) ·discretization rate
e
(10)
Binary search has only O(log
2
|S|) complexity
where |S| is the size of the (score; P-value) list. By
default we calculate this list sampling the score dis-
tributions in several points such that P-value differs
by no more than a given relative error δ for adjacent
pairs. The list of P-values is bounded by the minimal
achievable P-value minAchievablePvalue and 1. Thus
|S| = −
ln(minAchievablePvalue)
ln(1 + precision)
(11)
It’s also possible to make threshold to P-value
conversion in O(1) by expanding a P-value list into
an array indexed by discretized thresholds. Such an
algorithm consumes a bit more memory and works
notably faster than binary search. However, practical
profit in total computation time could be limited due
BIOINFORMATICS2015-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
104

to the score computation bottleneck. In turn, there is
room for score computation improvements using ad-
vanced methods (Korhonen et al., 2009).
4 RESULTS & DISCUSSION
4.1 Software Implementation
Our implementation of PERFECTOS-APE can be
used to test a set of SNVs against a given collec-
tion of TFBS models. PERFECTOS-APE is imple-
mented in Java and supports both mononucleotide and
dinucleotide PWMs. Console and web-based ver-
sions are available at the PERFECTOS-APE website
http://opera.autosome.ru/perfectosape/. Current im-
plementation is restricted to SNVs with two alter-
native alleles. PERFECTOS-APE web interface al-
lows testing a set of SNVs against publicly available
motif collections: HOCOMOCO (Kulakovskiy et al.,
2013b), JASPAR (Portales-Casamar et al., 2009), HT-
SELEX (Jolma et al., 2010), SwissRegulon (Pachkov
et al., 2007), and HOMER (Heinz et al., 2010).
We performed a basic benchmark with 100 SNPs
and the HOCOMOCO collection of 426 mononu-
cleotide PWMs on a common laptop with a Core
i3 CPU. Precalculation with default settings took ∼
150 Mb RAM and ∼ 30 seconds; the precalculation
step took ∼ 0.5 hour for a comparable amount of
dinucleotide PWMs. In the binary search mode pro-
cessing of 100 SNPs took ∼ 5 min/∼ 0.5 min and ∼
90Mb/∼ 200Mb RAM for basic/binary search mode
respectively. For dinucleotide motifs the running time
was ∼3 hours in basic mode and the same ∼0.5 min
as for the mononucleotide case was necessary in bi-
nary search mode. Thus, PERFECTOS-APE allows
large-scale SNP analysis.
4.2 Case Studies on Real Data
To test whether PERFECTOS-APE is able to produce
meaningful predictions we analysed several disease-
associated SNPs.
For instance, rSNP rs1314913 (Orr et al., 2012)
is located in an intronic region of RAD51B gene that
is significantly associated with breast cancer. It was
proposed (Orr et al., 2012) that the minor allele neg-
atively affects the binding site of the AP-1 complex.
PERFECTOS-APE reproduces this prediction (fig.1):
it shows 40× to 280× fold changes for binding site
P-values.
We also checked breast cancer-associated rSNPs,
which currently have no functional annotation (see
fig.2):
• rs3112612 (Fletcher et al., 2011) possibly dam-
ages the E4F1 binding site, and
• rs4784227 (Long et al., 2010) possibly damages
the HIC1 binding site.
Both those SNPs are located in the upstream region of
the TOX3 gene, thus possibly affecting not the protein
product directly, but its expression regulation.
To demonstrate PERFECTOS-APE performance
on genomic scale, we’ve taken data on somatic mu-
tations in 21 breast cancers (Nik-Zainal et al., 2012)
selecting only those in intronic or promoter regions
(76594 SNVs). It is known (Ostrow et al., 2014)
that cancer somatic mutations escape the pressure of
stabilizing selection responsible for maintaining ad-
equate tissue specific regulation of gene expression,
but fall under strong positive selection responsible
for fast cell division and survival in the tumor envi-
ronment. One of the sources of selection is disrup-
tion/emergence of a TFBS binding site for the mu-
tated allele. Two prevalent types of breast cancer so-
matic mutations, CpG and TpC with nucleotide sub-
stitutions at C, were considered independently.
Using 426 HOCOMOCO PWMs we predicted
TFBS overlapping the set of SNVs and passing
0.0005 P-value threshold for either the reference or
the mutated allele. Using PERFECTOS-APE we cal-
culated the number of cases when a somatic mutation
caused a significant negative TFBS affinity change for
the mutant allele (with a P-value cutoff at 5). To gen-
erate control data we shufflled 30bp sequences flank-
ing the SNVs (preserving 1bp mutation context) and
repeated TFBS prediction. This procedure was exe-
cuted 8 times aggregating the results obtained for the
same TF in the same mutation context, i.e. only CpG-
and TpC-context variants were considered for shuf-
fled sequences.
To evaluate whether somatic mutations are likely
to occur in the TFBS favoring context we have con-
structed 2x2 contingency tables for the number of
TFBS with a high/low affinity change caused by mu-
tations (as table columns) and genuine/shuffled SNV
context (as table rows). Fisher’s exact test was used
to estimate the significance of the association between
the significant change of TFBS affinity and the intact
SNV context. Since we tested all 426 HOCOMOCO
models for each mutation site, we calculated Holm’s
multiple testing correction for the number of TFBS
models.
We have found that original ”genuine” mutations
were the subject of either emergence or disruption
of TF binding sites significantly more often than the
shuffled random sequences.
In addition to Fisher’s Test Holm-corrected
P-values passing 0.05 threshold, we selected TFs for
PERFECTOS-APE-PredictingRegulatoryFunctionalEffectofSNPsbyApproximateP-valueEstimation
105

which the rate of TFBS disruption in real somatic mu-
tations was no less than 1.3 times greater than for the
synthetic data. The resulting list of TFs contained
several well-known oncogenes, such as HIF1A, SP2,
MYC for CpG context mutations and HLF, C/EBP-
family, RUNX2 and others for TpC context muta-
tions.
It is also notable, that a simpler approach compar-
ing overall counts of TFBS predictions overlapping
real somatic and ”shuffled” mutations was also show-
ing high significance (FDR-corrected Fisher’s exact
test P-value < 0.05) for the most of those TFs.
4.3 Comparison to Existing Tools
Existing tools for regulatory SNP annotation are
mostly provided as web services rather than stand-
alone command-line applications; the notable excep-
tions are sTRAP (implemented as R-package) and
FunSeq (a C++ based command line tool). Web-only
realization restricts applicability to large-scale data
sets.
Another limitation comes from predefined sets of
transcription factor binding motifs collections with
most of the web-tools providing only two built-in col-
lections: TRANSFAC and JASPAR. The choice of an
appropriate motif collection plays an important role in
finding effects of regulatory substitutions. It is desir-
able to have an option to utilize a user-defined motif
collection since the general purpose databases are in-
complete and motifs for new TFs keep emerging.
P-value ratio is a common approximation for bind-
ing affinity change and is used by almost all tools
except RAVEN (Andersen et al., 2008), which cal-
culates score difference. PERFECTOS-APE uses an
approach for P-value calculation very similar to that
used in is-rSNP (Macintyre et al., 2010); it allows
exact calculations of P-value with a given precision.
This differs our approach from RegSNP (Teng et al.,
2012) and sTRAP (Manke et al., 2010) which rely on
approximate techniques.
Most of tools use PWM motif models whereas
PERFECTOS-APE supports both PWM and diPWM.
The other tool that employs motif models more com-
plex than PWMs is rSNP-MAPPER, which uses Hid-
den Markov Models.
RAVEN, ChroMoS (Barenboim and Manke,
2013), RegSNP and FunSeq (Khurana et al., 2013)
are complex integrative tools which provide an-
notation compiling information from different data
sources, while is-rSNP, sTRAP, rSNP-Mapper (Riva,
2012) and PERFECTOS-APE perform solely se-
quence analysis.
PERFECTOS-APE was intentionally designed as
a stand-alone tool accepting raw sequences (not gene
names or dbSNP identifiers) and making no prelim-
inary filtering of SNPs according to their location,
functionality etc. (unlike integrative tools).
PERFECTOS-APE does not perform any multi-
ple testing correction, which is left up to downstream
analysis if necessary.
We tested several tools on a verified
SNP (rs1314913) and gathered ranks of AP-1
complex in the resulting predictions (see table 1).
Our observations show that all methods behave sim-
ilarly, but the results depend on the motif collection.
For instance, is-rSNP fails to recognize rs1314913
as a regulatory SNP using JASPAR motif collection.
For other tools ranks of transcription factors vary
significantly depending on selected collection.
4.4 Discussion
PERFECTOS-APE can be improved in many ways,
including direct support for multiallelic SNVs and
more efficient procedure to scan sequences for bind-
ing sites. Also an effective parallelization is possi-
ble since each SNV versus PWM test can be run as
a separate task or thread. Dinucleotide PWMs have
shown better TFBS recognition than classic PWMs
(Kulakovskiy et al., 2013a; Levitsky et al., 2014), yet
the classic PWMs are much more common and are
available for a wider range of transcription factors.
As more data on TF binding becomes available and
advanced models are constructed for a wider range
of TFs, PERFECTOS-APE can be efficiently used for
analysis of regulatory SNVs and SNPs.
ACKNOWLEDGMENTS
The authors thank the School for Molecular and The-
oretical Biology of the Dynasty Foundation, and per-
sonally Anya Piotrovskaya and Fyodor Kondrashov.
This work was supported by the Dynasty Foundation
Fellowship; by Russian Scientific Foundation [14-14-
01140]; by the Program on Molecular and Cell Biol-
ogy from the Presidium of RAS.
REFERENCES
Andersen, M. C., Engstr
¨
om, P. G., Lithwick, S., Arenillas,
D., Eriksson, P., Lenhard, B., Wasserman, W. W., and
Odeberg, J. (2008). In silico detection of sequence
variations modifying transcriptional regulation. PLoS
computational biology, 4(1):e5.
BIOINFORMATICS2015-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
106

Barenboim, M. and Manke, T. (2013). Chromos: an in-
tegrated web tool for snp classification, prioritiza-
tion and functional interpretation. Bioinformatics,
29(17):2197–2198.
Fletcher, O., Johnson, N., Orr, N., Hosking, F. J., Gib-
son, L. J., Walker, K., Zelenika, D., Gut, I., Heath,
S., Palles, C., et al. (2011). Novel breast cancer sus-
ceptibility locus at 9q31. 2: results of a genome-wide
association study. Journal of the National Cancer In-
stitute.
Heinz, S., Benner, C., Spann, N., Bertolino, E., Lin, Y. C.,
Laslo, P., Cheng, J. X., Murre, C., Singh, H., and
Glass, C. K. (2010). Simple combinations of lineage-
determining transcription factors prime cis-regulatory
elements required for macrophage and b cell identi-
ties. Molecular cell, 38(4):576–589.
Jolma, A., Kivioja, T., Toivonen, J., Cheng, L., Wei, G.,
Enge, M., Taipale, M., Vaquerizas, J. M., Yan, J., Sil-
lanp
¨
a
¨
a, M. J., et al. (2010). Multiplexed massively
parallel selex for characterization of human transcrip-
tion factor binding specificities. Genome research,
20(6):861–873.
Khurana, E., Fu, Y., Colonna, V., Mu, X. J., Kang, H. M.,
Lappalainen, T., Sboner, A., Lochovsky, L., Chen, J.,
Harmanci, A., et al. (2013). Integrative annotation of
variants from 1092 humans: Application to cancer ge-
nomics. Science, 342(6154):1235587.
Korhonen, J., Martinm
¨
aki, P., Pizzi, C., Rastas, P., and
Ukkonen, E. (2009). Moods: fast search for position
weight matrix matches in dna sequences. Bioinfor-
matics, 25(23):3181–3182.
Kulakovskiy, I., Levitsky, V., Oshchepkov, D., Bryzgalov,
L., Vorontsov, I., and Makeev, V. (2013a). From bind-
ing motifs in chip-seq data to improved models of
transcription factor binding sites. Journal of bioin-
formatics and computational biology, 11(01).
Kulakovskiy, I. V., Medvedeva, Y. A., Schaefer, U.,
Kasianov, A. S., Vorontsov, I. E., Bajic, V. B., and
Makeev, V. J. (2013b). Hocomoco: a comprehensive
collection of human transcription factor binding sites
models. Nucleic acids research, 41(D1):D195–D202.
Levitsky, V. G., Kulakovskiy, I. V., Ershov, N. I., Os-
chepkov, D. Y., Makeev, V. J., Hodgman, T., and
Merkulova, T. I. (2014). Application of experimen-
tally verified transcription factor binding sites models
for computational analysis of chip-seq data. BMC ge-
nomics, 15(1):80.
Long, J., Cai, Q., Shu, X.-O., Qu, S., Li, C., Zheng, Y., Gu,
K., Wang, W., Xiang, Y.-B., Cheng, J., et al. (2010).
Identification of a functional genetic variant at 16q12.
1 for breast cancer risk: results from the asia breast
cancer consortium. PLoS genetics, 6(6):e1001002.
Macintyre, G., Bailey, J., Haviv, I., and Kowalczyk, A.
(2010). is-rsnp: a novel technique for in silico regula-
tory snp detection. Bioinformatics, 26(18):i524–i530.
Manke, T., Heinig, M., and Vingron, M. (2010). Quan-
tifying the effect of sequence variation on regulatory
interactions. Human mutation, 31(4):477–483.
Mathelier, A. and Wasserman, W. W. (2013). The next gen-
eration of transcription factor binding site prediction.
PLoS computational biology, 9(9):e1003214.
Nik-Zainal, S., Alexandrov, L. B., Wedge, D. C., Van Loo,
P., Greenman, C. D., Raine, K., Jones, D., Hinton,
J., Marshall, J., Stebbings, L. A., et al. (2012). Mu-
tational processes molding the genomes of 21 breast
cancers. Cell, 149(5):979–993.
Orr, N., Lemnrau, A., Cooke, R., Fletcher, O., Tomczyk, K.,
Jones, M., Johnson, N., Lord, C. J., Mitsopoulos, C.,
Zvelebil, M., et al. (2012). Genome-wide association
study identifies a common variant in rad51b associ-
ated with male breast cancer risk. Nature genetics,
44(11):1182–1184.
Ostrow, S. L., Barshir, R., DeGregori, J., Yeger-Lotem, E.,
and Hershberg, R. (2014). Cancer evolution is associ-
ated with pervasive positive selection on globally ex-
pressed genes. PLoS genetics, 10(3):e1004239.
Pachkov, M., Erb, I., Molina, N., and Van Nimwegen, E.
(2007). Swissregulon: a database of genome-wide an-
notations of regulatory sites. Nucleic acids research,
35(suppl 1):D127–D131.
Ponomarenko, J. V., Merkulova, T. I., Vasiliev, G. V., Lev-
ashova, Z. B., Orlova, G. V., Lavryushev, S. V., Fokin,
O. N., Ponomarenko, M. P., Frolov, A. S., and Sarai,
A. (2001). rsnp guide, a database system for analysis
of transcription factor binding to target sequences: ap-
plication to snps and site-directed mutations. Nucleic
acids research, 29(1):312–316.
Portales-Casamar, E., Thongjuea, S., Kwon, A. T., Arenil-
las, D., Zhao, X., Valen, E., Yusuf, D., Lenhard, B.,
Wasserman, W. W., and Sandelin, A. (2009). Jaspar
2010: the greatly expanded open-access database of
transcription factor binding profiles. Nucleic acids re-
search, page gkp950.
Riva, A. (2012). Large-scale computational identification
of regulatory snps with rsnp-mapper. BMC genomics,
13(Suppl 4):S7.
Teng, M., Ichikawa, S., Padgett, L. R., Wang, Y., Mort, M.,
Cooper, D. N., Koller, D. L., Foroud, T., Edenberg,
H. J., Econs, M. J., et al. (2012). regsnps: a strategy
for prioritizing regulatory single nucleotide substitu-
tions. Bioinformatics, 28(14):1879–1886.
Touzet, H., Varr
´
e, J.-S., et al. (2007). Efficient and accurate
p-value computation for position weight matrices. Al-
gorithms Mol Biol, 2(1510.1186):1748–7188.
PERFECTOS-APE-PredictingRegulatoryFunctionalEffectofSNPsbyApproximateP-valueEstimation
107
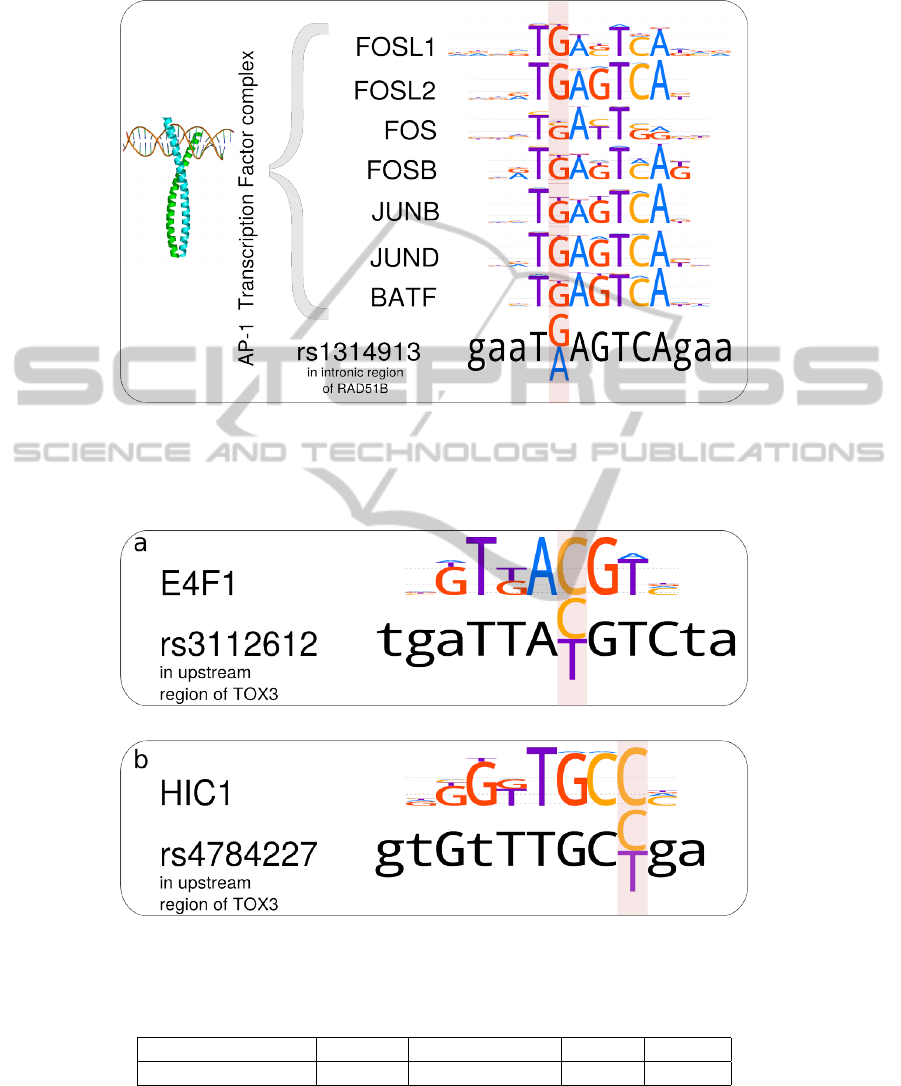
APPENDIX
Figure 1: SNP rs1314913 is located in the intronic region of the breast cancer-associated RAD51B gene that is involved
in DNA recombination and DNA repair. The T(A) allele is associated with about 1.6 times higher odds of breast cancer
in men (Orr et al., 2012). Motif LOGO representations are given for HOCOMOCO PWMs (Kulakovskiy et al., 2013b).
PERFECTOS-APE P-value fold changes are 40× to 280×, TFBS P-value threshold was 0.0005
Figure 2: Breast cancer-associated SNPs in the promoter region of the TOX3 gene (fold-change cutoff 5; TFBS P-value
threshold 0.001). (a) rs3112612, Risk allele (T) damages putative E4F1 binding site. (b) rs4784227, Risk allele (T) damages
putative HIC1 binding site.
Table 1: AP-1 complex ranking in resulting predictions for rs1314913 analyzed by different tools.
PERFECTOS-APE RegSNP rSNP-MAPPER sTRAP is-rSNP
1-7,13 9,10,13 3,5,6,9 1-6 3,8
BIOINFORMATICS2015-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
108
