
Is the Identification of SNP-miRNA Interactions Supporting the
Prediction of Human Lymphocyte Transcriptional Radiation Responses?
Marzena Dolbniak
1
, Joanna Zyla
1
, Sylwia Kabacik
2
, Grainne Manning
2
, Christophe Badie
2
,
Ghazi Alsbeih
3
and Joanna Polanska
1
1
Institute of Automatic Control, Faculty of Automatic Control, Electronic and Computer Science,
Silesian University of Technology, Akademicka 16, Gliwice, Poland
2
Cancer Genetics and Cytogenetics Group, Biological Effects Department,
Centre for Radiation, Chemical and Environmental Hazards, Public Health England, Didcot, OX11 ORQ, U.K.
3
Radiation Biology Section, Biomedical Physics Dept., King Faisal Specialist Hospital & Research Centre,
Riyadh 11211, Kingdom of Saudi Arabia
Keywords:
GWAS, miRNA, Single Nucleotide Polymorphism, Radiosensitivity, Gene Expression, Radiation.
Abstract:
Genome-Wide Association Studies (GWAS) are of great importance in identifying the genetic variants asso-
ciated with traits/diseases. Due to the high number of candidate SNPs some filtering techniques are necessary
to be applied. The aim of the study was to develop the comprehensive approach allowing for detailed analysis
of both SNP-gene and SNP-miRNA-gene relations. We elaborated and optimized the novel signal analysis
pipeline improving significantly the results of the analysis on genotype-phenotype interplay. Direct links be-
tween genotype results and gene expression levels were enriched by detailed analysis of SNP-miRNA-gene
interactions at both mature miRNA structure/seed region and target binding site level. The proposed technique
was applied to the data on lymphocyte radiation response and increased by almost 100% number of potential
functional SNPs.
1 INTRODUCTION
Genome-Wide Association Studies (GWAS) are the
most popular kind of research to identify the ge-
netic variants associated with traits/diseases. Meth-
ods to identify the candidate single nucleotide poly-
morphisms (SNPs) depend on the study design and
different statistical approaches and have been widely
discussed in (Bush and Moore, 2012; Evangelou and
Ioannidis, 2013). Filtering the obtained set of can-
didate SNPs by applying the biological information,
such as the potential effects of mutation in gene cod-
ing regions, modifications of gene related signal path-
ways or gene ontology terms, or the detailed analysis
of microRNA-SNP interactions can reduce the num-
ber of false candidate SNPs, and become a separate
research field in GWAS analysis. Over the years, a
lot of algorithms were proposed to support such func-
tional analysis in (Patnala et al., 2013; Wang et al.,
2010), but the most challenging issue still belongs to
the prediction of microRNA-SNP interactions, as the
understanding of microRNAs function continue to in-
crease.
MicroRNAs (miRNAs) are small non-coding
RNAs, which regulate gene expression. Single nu-
cleotide polymorphisms may be functional with re-
spect to miRNAs biogenesis or the specific roles of
mature miRNA (Dong et al., 2013). From the miR-
NAs biogenesis pathway, SNPs can modify the pri-
mary miRNA (pre-miRNA) or mature miRNA struc-
tures. While considering the regulatory function of
miRNA, specifically relevant target site recognition
of the seed region of miRNA (6-8 nucleotides at
the 5’ end of the miRNA) is of great importance.
Roughly, single nucleotide polymorphisms can im-
pact the functionality of miRNA by modifying its pri-
mary target binding sites or by creating new binding
sites. As a consequence gene disregulation may lead
to phenotype changes and eventually prove to be crit-
ical for the susceptibility to cancer as well as other
diseases (Slaby et al., 2012; Vitale et al., 2011). Al-
though some of the published works include miRNA-
SNP interaction analysis, most of them focus on SNPs
in primary binding site only (Deveci et al., 2014).
Radiation sensitivity (radiosensitivity), which is
the relative susceptibility of cells, tissues, organs or
243
Dolbniak M., Zyla J., Kabacik S., Manning G., Badie C., Alsbeih G. and Polanska J..
Is the Identification of SNP-miRNA Interactions Supporting the Prediction of Human Lymphocyte Transcriptional Radiation Responses?.
DOI: 10.5220/0005286102430250
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2015), pages 243-250
ISBN: 978-989-758-070-3
Copyright
c
2015 SCITEPRESS (Science and Technology Publications, Lda.)

organisms to the harmful effect of radiation, can be
influenced by many factors amongst which are epi-
genetic modifications (Ma et al., 2010; Lahtz and
Pfeifer, 2011), and miRNA regulation (Zhao et al.,
2012). Among the group of radiation-responsive
genes, BBC3 (BCL2 binding component 3) is of great
importance, since it encodes a protein named PUMA
(p53 upregulated modulator of apoptosis) which is in-
volved in p53-dependent and -independent apoptosis
induced by a variety of signals amongst which there
is ionizing radiation (Yu and Zhang, 2005). This gene
is known as a reliable biomarker of radiation expo-
sure (Budworth et al., 2012). It is a good candi-
date to investigate the potential role of SNP-gene and
SNP-miRNA interactions in radiosensitivity and on
the long term, relevant for a better understanding of
inter-individual radiation responsiveness supposed to
be linked at least partially to apoptosis mechanisms.
This could lead to radiotherapy regime improvements
personalized cancer treatments.
In this study we perform a novel comprehen-
sive functional analysis taking into consideration both
SNP-gene and SNP-miRNA interactions. We demon-
strate that this type of approach can potentially im-
prove the discovery of candidate process relevant
SNPs compared to standard SNP-gene based methods
only.
2 MATERIALS AND METHODS
2.1 Materials
The group under investigation is composed of 44 un-
related Caucasian individuals (unR), with two types
of data collected.
The first dataset includes qPCR measurements for
BBC3 gene, taken in two conditions: 1) in normal
conditions - no irradiation, and 2) just after the irradi-
ation with a single dose of 2Gy. The irradiation was
performed at room temperature with an A.G.O. HS
X-ray system (Aldermaston, Reading, UK) (output 13
mA, 250 kV peak, 0.5 Gy/min for doses 0.5 4 Gy and
0.2 mA 4.9 mGy/min for doses up to 100 mGy). The
T-lymphocyte cultures were used and prepared using
the method described previously (O’Donovan et al.,
1995; Finnon et al., 2008).
The second dataset includes results on genotyp-
ing 567,096 polymorphisms (SNP) by Axiom GW
Human hg36.1 arrays. The BBC3 gene expressions
obtained at both experimental conditions were previ-
ously published (Kabacik et al., 2011a; Kabacik et al.,
2011b; Manning et al., 2013).
2.2 Selection of Polymorphisms
At the first step, the quality control for both qPCR
and microarray experiments was performed. Dur-
ing the next step, the genotype-phenotype interac-
tions were modelled per every SNP following the pro-
cedure previously presented in (Zyla et al., 2014).
The genotype-phenotype models were constructed for
both BBC3 expression level in normal condition (no
irradiation, 0Gy) and for standardized fold change
(FCH) signal. The final two sets of candidate ra-
diosensitivity related SNPs were defined as follows:
1) all SNPs significantly (p-value <0.05) related to
the BBC3 gene expression fold change (FCH) and
not significantly related to the BBC3 gene expression
level in normal condition (0Gy) - named SET 1, and
2) all SNPs significantly related to BBC3 gene ex-
pression fold change (FCH) and significantly related
to the BBC3 gene expression level in normal condi-
tion (0Gy) - SET 2 (Figure 1).
Figure 1: The definition of the final two sets of candidate
radiosensitivity related SNPs: 1) SET 1 - all SNPs signifi-
cantly related to the expression fold change (FCH) and not
significantly related to the expression in normal condition
(0Gy) - dark grey colour; 2) SET 2 - all SNPs significantly
related to the expression fold change (FCH) and signifi-
cantly related to the expression in normal condition (0Gy) -
light grey colour.
2.3 Functional Analysis
2.3.1 SNP-gene Interactions
The obtained two sets of candidate radiosensitivity
related SNPs were limited to the SNPs occurring in
genes. The information on SNP location in genome
and its transcriptomic assignment was collected us-
ing SNPLab software (GRCh38). Each SNP located
in exon and having missense activity (nonsynony-
mous SNP - nsSNPs) was further analysed to pre-
dict its impact on cell functioning. Polymorphisms
of this type lead to a change of the amino acid in the
protein sequence. To assess the impact of nsSNPs
on the organism the PredictSNP software was used
(Bendl et al., 2014), which integrates the results from
six the most popular algorithms (MAPP, SIFT SNAP,
BIOINFORMATICS2015-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
244
PolyPhen1, PolyPhen2, and PhD-SNP). Additionally,
each of the genes modified by candidate SNPs was in-
vestigated toward the overrepresentation of Gene On-
tology (GO) terms (Szkiba et al., 2014), and KEGG
pathways (Beissbarth and Speed, 2004).
2.3.2 SNP-miRNA interactions - standard
approach
Three the most popular bioinformatical systems
which analyse miRNA and SNP interactions are:
1) SNPinfo (Xu and Taylor, 2009), 2) PolymiRTS
Database (Bhattacharya et al., 2014), and 3) miRSNP
(Liu et al., 2012). All of the above focus mainly
on SNP and miRNA binding site interactions, in
particular SNPinfo focuses on polymorphisms in 3’
UTR region of mRNA and uses miRanda algorithm
(Griffiths-Jones et al., 2008) for target binding site
prediction, while PolymiRTS Database searches for
SNPs modifying seed regions of miRNA and uses
TargetScan algorithm (Lewis et al., 2005) and experi-
mental data (Dweep et al., 2014; Hsu et al., 2014; Ver-
goulis et al., 2012) for target binding site predictions.
The last of the mentioned algorithms, miRSNP, as the
only one identifies polymorphisms in pre-miRNA se-
quences and uses miRanda algorithm for identifica-
tion of SNPs modifying the binding sites.
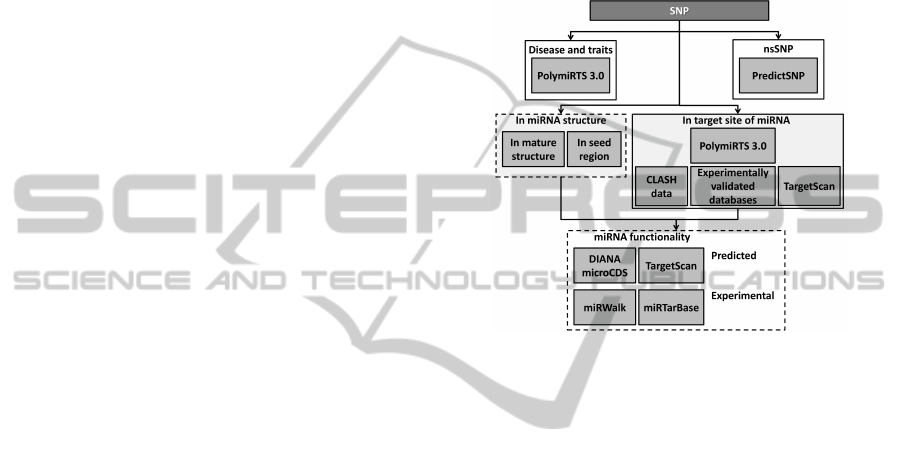
2.3.3 Comprehensive SNP-miRNA Analysis
Procedure
The detailed up-to-date analysis of SNP interaction on
mature miRNA or miRNA’s seed region done together
with tracking of binding site modifications seem to be
crucial for our study. Since none of the above sys-
tems allows for complete analysis of SNPs and mature
miRNA structure interactions, we have developed a
software performing the analyses. Using miRBase
v21 we found chromosomal coordinates (GRCh38) of
mature miRNAs and miRNAs seed regions and com-
pared them with coordinates of analysed SNPs from
previously found SET 1 or SET 2. Combining that
set with miRNAs having binding sites modified by
SNPs from the same SET 1 or SET 2 gives the com-
plete set of miRNAs under investigation allowing for
the detection of candidate SNPs responsible for mod-
ification of miRNA gene expression regulation pro-
cesses. The next step required the definition of bind-
ing sites being targets for chosen miRNAs, and it was
done at two levels: experimentally validated sites and
in silico predicted sites only. We used miRTarBase
(Hsu et al., 2014) and miRWalk (Dweep et al., 2014)
to find experimentally validated targets and for every
miRNA which does not have validated target genes
we used TargetScan and DIANA-microT-CDS predic-
tion algorithms. As it has become a common practice
for researches to look at predictions produces by sev-
eral miRNA-target prediction programs, we focused
on intersection of results obtained from this two algo-
rithms. The group of genes being targets for chosen
miRNAs was investigated toward the overrepresenta-
tion of Gene Ontology (GO) terms and KEGG path-
ways (Beissbarth and Speed, 2004). The data flow
pipeline for whole procedure is presented in Figure 2.
Figure 2: Scheme of data flow in SNP-miRNA interaction
identification.
3 RESULTS AND DISCUSSION
Quality control revealed that one of the SNPs was
missing in 91% cases and it was removed from fur-
ther analysis. None outliers were detected in qPCRs.
The total of 472,712 SNPs was considered during
the BBC3 gene expression interaction modelling due
to the lack of diveristy in the analysed samples for
94,383 SNPs. Table 1 includes the number of SNPs
representing three types of modelled interactions be-
tween BBC3 gene expression level and genotyped
polymorphisms (genotype, dominant and recessive).
For both endpoints (0Gy and FCH) number of SNPs
with particular interaction model being minimum p-
value was given in the row named Best model chosen,
while the next row gives the number of SNPs signif-
icant at α=0.05, split by SNP-BBC3 gene interaction
model type.
While analysing the expression of BBC3 gene,
the majority of SNPs represent dominant or recessive
model of interaction with FDR level around 50%. All
the polymorphisms with minimal p-value being less
than 0.05 were considered as the candidate SNPs re-
lated to the radiosensitivity phenomena. After apply-
ing the inclusion criteria defined for the final two sets
IstheIdentificationofSNP-miRNAInteractionsSupportingthePredictionofHumanLymphocyteTranscriptional
RadiationResponses?
245

Table 1: The results of the model selection for BBC3 at both
endpoints - 0Gy and FCH.
G D R TOTAL
Total 183,965 309,155 348,188 841,308
0Gy
p<0.05 9,614 37,672 42,559 89,845
FDR [%] 95.68 41.03 40.91 46.81
FCH
p<0.05 8,833 31,084 34,440 74,357
FDR [%] 100 49.73 50.55 56.57
0Gy
Best model 2,462 215,268 254,930 472,712
p<0.05 1,550 35,073 39,865 76,488
FCH
Best model 2,670 215,590 254,452 472,712
p<0.05 1,525 29,034 32,355 62,914
G - Genotype model; D - Dominant model; R - Recessive model.
of candidate radiosensitivity related SNPs, SET 1 in-
cluded 40,953 SNPs, while SET 2 consists of 21,961
SNPs.
3.1 Functional Analysis
3.1.1 SNP-gene Interactions
Table 2 presents detailed information about function-
ality of significant SNPs at two investigated end-
points.
Table 2: Transcriptomic location of candidate SNPs SET 1
and SET 2.
No. of SNPs in particular transcriptomic location
SET 1 [%] SET 2 [%]
TOTAL 40,953 100 21,961 100
Total functional 17,096 41.8 9,169 41.8
intron 15,413 37.6 8,271 37.7
exon
synSNP 105 0.3 69 0.3
nsSNP 244 0.6 121 0.6
UTR3’ 418 1.0 223 1.0
UTR5’ 67 0.2 29 0.1
nearGene3’ * 83 0.2 34 0.2
nearGene5’ ** 287 0.7 171 0.8
splice3’ 1 0.01 0 0.0
splice5’ 5 0.01 1 0.01
frameshift 1 0.01 0 0.0
STOP codon loss 1 0.01 0 0.0
ncRNA 94 0.2 53 0.2
cds-reference 377 0.9 197 0.9
* nearGene3’ - within 3’ 0.5kb to a gene;
** nearGene5’ - within 5’ 2kb to a gene.
Initially, the impact of nonsynonymousSNP
(nsSNP) was investigated by PredictSNP software
and the results are presented in table 3. For all dele-
terious SNPs literature study was performed and for
SET 1 three of them occur in genes highly relevant
and affiliated to cancer processes: AMACR (Jianq
et al., 2013), SERPINB5 (Kapoor, 2014), ABCC11
(Yamada et al., 2013): In SET 2 only one gene is
highly relevant to cancer processes: TLR6 (Miedema
et al., 2012).
Table 3: The percentage of deleterious nsSNPs in SET 1
and SET 2 candidate SNPs.
nsSNP predictions
SET 1 SET 2
Total 244 122
Deleterious 27 20
[%] 11.07 16.39
Additionally, both groups of genes with sig-
nificant functional SNP-gene interactions, equal to
5,450 in case of SET 1 and 3,505 for SET
2, were investigated on the overrepresentation of
KEGG pathways and the summary of that analy-
sis presents table 4. In general, 45 KEGG path-
ways are overrepresented either in genes related to
SET 1 or SET 2. Twenty two of them (48.89%)
are significantly overrepresented in both gene sets.
Among 32 significantly overrepresented KEGG path-
ways for genes disregulated by candidate func-
tional SNPs from SET 2, the highest odds ratio
was noticed for ko00604 (Glycosphingolipid biosyn-
thesis, OR=4.51, p-value=0.010638) and ko00532
(Glycosaminoglycan biosynthesis, OR=3.68, p-
value=0.012608) which is consistent with the latest
literature based reports on the strong relation between
these two processes and irradiation or cancerogene-
sis (Aureli et al., 2014; Hirshoren et al., 2014). One
of the most significant overrepresented pathways was
ko04360 (Axon guidance, OR=2.97, p-value=3.32E-
07), process strongly related to local invasion and
metastatic spread of the tumour (Ochi et al., 2002).
All these three pathways were also significantly over-
represented among genes modified by SNPs from
SET 1 (p-values equal to 0.027915, 0.003554, and
2.75E-07 for ko00604, ko00532, and ko04360 respec-
tively). While looking at the signal pathways differen-
tially represented in both gene sets, ko04810 (Regula-
tion of actin cytoskeleton), ko04520 (Adherens junc-
tion), and ko05200 (Pathways in cancer) are overrep-
resented in SET 1 - related to the response to irradi-
ation with no relation to 0Gy, and are not enriched
in SET 2, which includes genes related to both to
background (0Gy response) and response to irradia-
tion level (FCH) (Bansal et al., 2014). The oppo-
site analysis - looking for pathways overrepresented
in SET 2 and not enriched in SET 1 brings ko04540
(Gap junction) and ko04210 (Apoptosis) known as re-
BIOINFORMATICS2015-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
246

lated to radioadaptive response (Nenoi et al., 2014).
The detailed analysis of pathways overrepresented
in both SET 1 and SET 2, three signal pathways,
highly relevant to cancerogenesis, are overrepresen-
taed in both groups of genes - SET 1 and SET
2. Calcium signalling pathway (ko04020) plays the
main role in cell signalling and is thought, for exam-
ple, to have an impact for preventing metastases in
breast cancer (Davis et al., 2014). MAPK signalling
(ko04010) is the main path responsible for cell com-
munication and reaction for stress. Many types of
cancers have mutations in genes in MAPK pathway
(Dhillon et al., 2007). The third one, focal adhesion
(ko04510) has recently been identified as key deter-
minat of cancer cell resistance to radio- and chemo-
therapy (Eke and Cordes, 2014).
Table 4: Details of KEGG pathways overrepresentation
analysis among genes modified by SET 1 and SET 2 candi-
date nsSNPs.
KEGG pathways
SET 1 SET 2
No. of SNPs 17,096 21,961
No. of genes 5,450 3,505
No. of analysed KEGG pathways 232 227
No. of overrepresented
KEGG pathways 35 32
FDR [%] 33.14 35.47
3.2 SNP-miRNA-gene Interactions
Summary of the results obtained presents table 5.
Novel comprehensive approach allows to identify
both types of interactions (polymorphism inside ma-
ture miRNAs and in target sites) which cannot be ob-
tained with the use of standard data analysis systems,
and which significantly increases the number of ob-
served interactions.
Table 5: Summary of the comprehensive SNP-miRNA in-
teraction analysis for two SNP sets SET 1 and SET 2.
No. of miRNA and polymorphisms interactions
SET 1 SET 2
SNPs in miRNA structure
mature miRNA 13 2
seed region 4 2
SNPs in target site of miRNA
CLASH 9 8
experimentally validated 2 2
in silico predicted 414 218
Several involved in cancer progression miRNAs,
with polymorphisms in mature structure were found.
The most relevant are: hsa-let-7a-3p with rs12326928
(p-value=0.0101; strong impact on breast cancer)
(Yu et al., 2007)), hsa-miR-519c-5p with rs1816087
(p-value=0.0101; regulate human breast cancer
resistance protein) (Li et al., 2011)), hsa-miR-512-3p
with rs4145874 (p-value=0.0119; regulation of genes
associated with cancer) (Chen et al., 2010)), and
hsa-miR-22-5p with rs9828426 (p-value=0.0482;
regulation of genes associated with breast cancer
(Patel et al., 2011)).
Exemplary polymorphism modifying mature
miRNA among those found for both SET 1 and SET
2 candidate SNP sets is rs2974617 (p-value=0.0152).
It modifies the mature structure of hsa-miR-4796-5p
and is located in TRIM36 gene. This gene and its
products are members of tripartite motif (TRIM)
family. Most of the genes from TRIM family are
observed being differentially expressed in many
types of cancers. The TRIM36 has a significant role
in chromosome segregation and cell cycle regulation
(Hatakeyamai, 2011). hsa-miR-4796-5p is not well
described in literature. However, we found 40
potential target genes. This group was investigated
by overrepresentation analysis of GO and KEGG
pathways. The most important gene ontology term is
GO:0097237 (cellular response to toxic substance;
p-value=0.038) and already mentioned ko04020
(calcium signalling pathway; p-value=0.015).
The parallel analysis was performed to find
significant polymorphisms located in miRNAs target
sites. While looking at the results obtained with the
use of CLASH database, SET 1 contains, for exam-
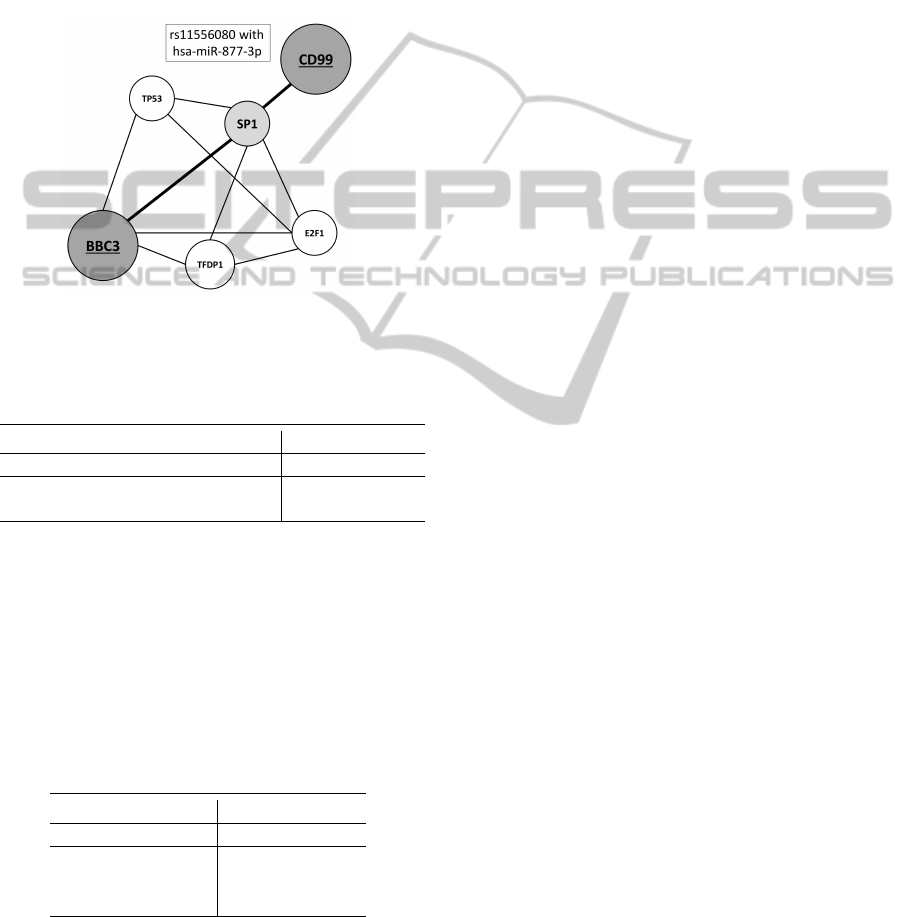
ple, polymorphism rs11556080 (p-value=0.0270),
which changes the regulation of CD99 gene. CD99
gene is located on X chromosome and unlikely to
most of genes, it does not undergo X inactivation.
CD99 is found in tumour cell of Ewing’s sarcoma.
Its knockdown reduces the tumour progression
(Rocchi et al., 2010). The recent studies suggest
CD99 as a biological marker for non-small lung
cancer (Edlund et al., 2012). This dysregulation can
be connected with the distribution of target site for
hsa-miR-877-3p. The results of DIANA software
(mirPath, 0.8 MicroT threshold) suggest that it can
interact with genes connected with cancer (ko05200
pathways in cancer; p-value=0.006) and ko04115
(p53 signaling pathway; p-value=0.017) - figure 3.
The SET 2 contains, among the others, rs989902
and rs184967 located respectively in PTPN13 and
MSH3 genes. The first polymorphism can disturb
target site for hsa-miR-186-5p (role in human colon
carcinoma cells (Chen et al., 2013)), the second
hsa-miR-92b-3p (connected with brain cancer and
metastasis (Nass et al., 2009)). Both genes are highly
IstheIdentificationofSNP-miRNAInteractionsSupportingthePredictionofHumanLymphocyteTranscriptional
RadiationResponses?
247
relevant for investigated trait. PTPN13 plays the
role in the process of metastasis in lung cancer (Han
et al., 2013), where MSH3 is one of the main genes
responsible for miss-match in the repair process.
Also, gene MSH3 was found as a candidate to
describe the radiosensitivity phenomena (Mangoni
et al., 2011). Using PolymiRTS Database 3.0 we
obtained the information on relations of significant
polymorphisms to different diseases (presented in
table 6).
Figure 3: Pathway interaction between BBC3 and dysregu-
lated gene CD99 based on genemania.org.
Table 6: Number of diseases and traits associated with poly-
morphisms from SET 1 and SET 2.
SET 1 SET 2
No. of diseases/traits interactions 283 127
No. of unique diseases/traits 144 77
No. of cancer disease interaction 21 5
The comparison between novel comprehensive
approach and SNPinfo based one reveals, that ap-
plying the detailed analysis of SNP-miRNA interac-
tions combined with the integrative PredictSNP algo-
rithm significantly increases the number of candidate
functionally validated SNPs by 222 SNPs for SET 1
(92.88%, from 239 to 461) and by 150 SNPs for SET
2 (96.00%, from 125 to 245) - table 7.
Table 7: Summary of novel functional analysis for SET 1
and SET 2.
SET 1 SET 2
TOTAL 461 245
Deleterious nsSNPs 27 20
SNPs in target site 421 223
SNPs in miRNA 13 2
4 CONCLUSIONS
We developed the novel comprehensive technique
improving significantly the results of the analysis
on genotype-phenotype interactions. Direct links
between genotype results and gene expression lev-
els were enriched by detailed analysis of SNP-
miRNA-gene interactions at both mature miRNA
structure/seed region and target binding site level.
The presented analysis can filter out non-functional
SNPs from extremely large number of relevant poly-
morphisms resulting of GWAS analysis.
The proposed technique was applied to the prob-
lem of searching for genetic signature of radiosen-
sitivity. Eight polymorphisms highly relevant to the
process of description of the radiosensitivity phenom-
ena were obtained, majority of them were indirectly
validated during the literature study
ACKNOWLEDGEMENTS
The authors would like to thank Dr. S. Majid, Ms.
N. Al-Harbi, Ms. S. Al-Qahtani for running the Ax-
iom Affymetrix platform, Paul Finnon for cell cul-
ture, Anna Krawczyk, the author of SNPLab soft-
ware, for her help in data collection. The work was
financially supported by NCN grant HARMONIA
4 DEC-2013/08/M/ST6/00924 (JP), the National In-
stitute for Health Research Centre for Research in
Public Health Protection at PHE (CB), the National
Science, Technology & Innovation Plan (NSTIP)
Project 11-BIO1429-20 (KFSHRC RAC# 2120 003)
(GA), SUT- BKM/524/ RAU1/2014/t.6 (MD), SUT-
BKM/524/ RAU1/2014/t.16 (JZ). Additionally, MD
and JZ are holders of scholarship DoktoRis- Schol-
arship program for Innovative Silesia. Calculations
were carried out using infrastructure of GeCONiI
(POIG.02.03.01-24-099/13).
DECLARATION OF INTEREST
The authors alone are responsible for the content and
writing of the paper.
REFERENCES
Aureli, M., Murdica, V., Loberto, N., Samarani, M.,
Prinetti, A., Bassi, R., and Sonnino, S. (2014). Explor-
ing the link between ceramide and ionizing radiation.
Glycoconj J., 31(6-7):449–459.
BIOINFORMATICS2015-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
248

Bansal, N., Mims, J., Kuremsky, J., Olex, A., Zhao, W., Yin,
L., Wani, R., Qian, J., Center, B., Marrs, G., Poros-
nicu, M., Fetrow, J., Tsang, A., and Furdui, C. (2014).
Broad phenotypic changes associated with gain of ra-
diation resistance in head and neck squamous cell can-
cer. Antioxid Redox Signal., 21(2):221–236.
Beissbarth, T. and Speed, T. (2004). GOstat: find statisti-
cally overrepresented gene ontologies within a group
of genes. Bioinformatics., 197(1):1464–1465.
Bendl, J., Stourac, J., Salanda, O., Pavelka, A., Wieben, E.,
Zendulka, J., Brezovsky, J., and Dombrsky, J. (2014).
PredictSNP: Robust and accurate consensus classi-
fier for prediction of disease-related mutations. PLoS
Comput Biol., 10(1):e1003440.
Bhattacharya, A., Ziebarth, J., and Cui, Y. (2014).
PolymiRTS database 3.0: linking polymorphisms in
microRNAs and their target sites with human dis-
eases and biological pathways. Nucleic Acids Res.,
42(D1):D86–D91.
Budworth, H., Snijders, A., Marchetti, F., Mannion, B.,
Bhatnagar, S., Kwoh, E., Tan, Y., Wang, S., Blakely,
W., Coleman, M., Peterson, L., and Wyrobek, A.
(2012). Dna repair and cell cycle biomarkers of ra-
diation exposure and inflammation stress in human
blood. PLoS One, 7(11):e48619.
Bush, W. and Moore, J. (2012). Chapter 11: Genome-wide
association studies. PLoS Comput Biol., 8(12).
Chen, F., Zhou, C., Lu, Y., Yuan, L., Peng, F., Zheng, L.,
and Li, X. (2013). Expression of hsa-mir-186 and its
role in human colon carcinoma cells. Nan Fang Yi Ke
Da Xue Xue Bao, 33(5):654–60.
Chen, F., Zhu, H., Zhou, L., Wu, S., Wang, J., and Chen, Z.
(2010). Inhibition of c-flip expression by mir-512-3p
contributes to taxol-induced apoptosis in hepatocellu-
lar carcinoma cells. Oncology Reports, 23(5):1457–
1462.
Davis, F., Azimi, I., Faville, R., Peters, A., Jalink, K.,
Putney, J., Goodhill, G., Thompson, E., Roberts-
Thomson, S., and Monteith, G. (2014). Induction
of epithelialmesenchymal transition (EMT) in breast
cancer cells is calcium signal dependent. Oncogene.,
33:2307–2316.
Deveci, M., Catalyrek, U., Svoboda, M., and Toland,
A. (2014). mrSNP: Software to detect SNP ef-
fects on microRNA binding. BMC Bioinformatics.,
15):doi:10.1186/1471–2105–15–73.
Dhillon, A., Hagan, S., Rath, O., and Kolc, W. (2007).
MAP kinase signalling pathways in cancer. Onco-
gene., 26:3279–3290.
Dong, H., Lei, J., Ding, L., Wen, Y., Ju, H., and Zhang, X.
(2013). MicroRNA: Function, detection, and bioanal-
ysis. Chem. Rev., 113(8):6207–6233.
Dweep, H., Sticht, C., Pandey, P., and Gretz, N. (2014).
miRWalk - database: prediction of possible mirna
binding sites by ’walking’ the genes of 3 genomes.
Nucleic Acids Research, 42:D78D85.
Edlund, K., Lindskog, C., Saito, A., Berglund, A., Pontn,
F., Gransson-Kultima, H., Isaksson, A., Jirstrm, K.,
Planck, M., Johansson, L., Lambe, M., Holmberg,
L., Nyberg, F., Ekman, S., Bergqvist, M., Lan-
delius, P., Lamberg, K., Botling, J., Ostman, A., and
Micke, P. (2012). CD99 is a novel prognostic stromal
marker in non-small cell lung cancer. Int J Cancer,
131(10):2264–2273.
Eke, I. and Cordes, N. (2014). Focal adhesion signaling and
therapy resistance in cancer. Semin Cancer Biol., pii:
S1044-579X(14)00098-4.
Evangelou, E. and Ioannidis, J. (2013). Meta-analysis meth-
ods for genome-wide association studies and beyond.
Nat. Rev. Genet., 14:379389.
Finnon, P., Robertson, N., Dziwura, D., Raffy, C., Zhang,
W., Ainsbury, L., Kaprio, J., Badie, C., and Bouf-
fler, S. (2008). Evidence for significant heritability
of apoptotic and cell cycle responses to ionising radi-
ation. Hum Genet., 123(5):485–493.
Griffiths-Jones, S., Saini, H., van Dongen, S., and Enright,
A. (2008). mirbase tools for microrna genomics. Nu-
cleic Acids, 36:154–158.
Han, X., Xue, L., Zhou, L., Gong, L., Zhu, S., Yao, L.,
Wang, S., Lan, M., Li, Y., and Zhang, W. (2013). The
role of ptpn13 in invasion and metastasis of lung squa-
mous cell carcinoma. Exp Mol Pathol., 95(3):270–58.
Hatakeyamai, S. (2011). TRIM proteins and cancer. Nat
Rev Cancer, 11(11):792–804.
Hirshoren, N., Bulvik, R., Neuman, T., Rubinstein, A.,
Meirovitz, A., and Elkin, M. (2014). Induction of hep-
aranase by HPV E6 oncogene in head and neck squa-
mous cell carcinoma. J Cell Mol Med., 18(1):181–
186.
Hsu, S., Tseng, Y., Shrestha, S., Lin, Y., Khaleel, A., Chou,
C., Chu, C., Huang, H., Lin, C., Ho, S., Jian, T.,
Lin, F., Chang, T., Weng, S., Liao, K., Liao, I., Liu,
C., and Huang, H. (2014). MiRTarBase update 2014:
an information resource for experimentally validated
miRNA-target interactions. Nucleic Acids Research,
42:D78D85.
Jianq, N., Zhu, S., Chen, J., Niu, Y., and Zhou, L. (2013).
A-methylacyl-CoA racemase (AMACR) and prostate-
cancer risk: a meta-analysis of 4,385 participants.
PLoS One., 8(10):e74386.
Kabacik, S., Mackay, A., Tamber, N., Manning, G., Finnon,
P., Paillier, F., Ashworth, A., Bouffler, S., and Badie,
C. (2011a). Gene expression following ionising radi-
ation: identification of biomarkers for dose estimation
and prediction of individual response. Int J Radiat
Biol., 87(2):115–129.
Kabacik, S., Ortega-Molina, A., Efayan, A., Finnon, P.,
Bouffler, S., Serrano, M., and Badie, C. (2011b). A
minimally invasive assay for individual assessment
of the atm/chek2/p53 pathway activity. Cell Cycle.,
10(7):1152–1161.
Kapoor, S. (2014). Maspin and its evolving role in tumor
progression in systemic malignancies. Breast Cancer.,
21(2):249.
Lahtz, C. and Pfeifer, G. (2011). Epigenetic changes of dna
repair genes in cancer. J Mol Cell Biol., 3(1):51–55.
Lewis, B., Burge, C., and Bartel, D. (2005). Conserved
seed pairing, often flanked by adenosines, indicates
IstheIdentificationofSNP-miRNAInteractionsSupportingthePredictionofHumanLymphocyteTranscriptional
RadiationResponses?
249

that thousands of human genes are microrna targets.
Cell, 120:15–20.
Li, X., Pan, Y., Seigel, G., Hu, Z., Huang, M., and
Yu, A. (2011). Breast cancer resistance protein
BCRP/ABCG2 regulatory microRNAs (hsa-mir-328,
-519c and -520h) and their differential expression in
stem-like ABCG2+ cancer cells. Biochemical Phar-
macology, 81(6):783 – 792.
Liu, C., Zhang, F., Li, T., Lu, M., Wang, L., Yue, W.,
and Zhang, D. (2012). MirSNP, a database of poly-
morphisms altering miRNA target sites, identifies
miRNA-related SNPs in GWAS SNPSs and eQTLs.
BMC Genomics., 13:661.
Ma, S., Liu, X., Jiao, B., Yang, Y., and Liu, X. (2010). Low-
dose radiation-induced responses: focusing on epige-
netic regulation. Int J Radiat Biol., 86(7):517–528.
Mangoni, M., Bisanzi, S., Carozzi, F., Sani, C., Biti, G.,
Livi, L., Barletta, E., Costantini, A., and Gorini,
G. (2011). Association between genetic polymor-
phisms in the xrcc1, xrcc3, xpd, gstm1, gstt1, msh2,
mlh1, msh3, and mgmt genes and radiosensitivity in
breast cancer patients. Int J Radiat Oncol Biol Phys.,
81(1):52–58.
Manning, G., Kabacik, S., Finnon, P., Bouffler, S., and
Badie, C. (2013). High and low dose responses of
transcriptional biomarkers in ex vivo x-irradiated hu-
man blood. Int J Radiat Biol., 89(7):511–522.
Miedema, K., Tissing, W., Poele, E. T., Kamps, W., Al-
izadeh, B., Kerkhof, M., de Jongste, J., Smit, H.,
de Pagter, A., Bierings, M., Boezen, H., Postma, D.,
de Bont, E., and Koppelman, G. (2012). Polymor-
phisms in the TLR6 gene associated with the inverse
association between childhood acute lymphoblastic
leukemia and atopic disease. Leukemia., 26(6):1203–
1210.
Nass, D., Rosenwald, S., ans S. Gilad, E. M., Tabibian-
Keissar, H., Schlosberg, A., Kuker, H., Sion-Vardy,
N., Tobar, A., Kharenko, O., Sitbon, E., Yanai, G. L.,
Elyakim, E., Cholakh, H., Gibori, H., Spector, Y.,
Bentwich, Z., Barshack, I., and Rosenfeld, N. (2009).
Mir-92b and mir-9/9* are specifically expressed in
brain primary tumors and can be used to differentiate
primary from metastatic brain tumors. Brain Pathol-
ogy, 19:375–383.
Nenoi, M., Wang, B., and Vares, G. (2014). In vivo ra-
dioadaptive response: A review of studies relevant to
radiation-induced cancer risk. Hum Exp Toxicol., pii:
0960327114537537.
Ochi, K., Mori, T., Toyama, Y., Nakamura, Y., and
Arakawa, H. (2002). Identification of semaphorin3B
as a direct target of p53. Neoplasia., 4(1):82–87.
O’Donovan, M., Freemantle, M., Hull, G., Bell, D., Arlett,
C., and Cole, J. (1995). Extended-term cultures of
human T-lymphocytes: a practical alternative to pri-
mary human lymphocytes for use in genotoxicity test-
ing. Mutagenesis., 10(3):189–201.
Patel, J., Appaiah, H., Burnett, R., Bhat-Nakshatri, P.,
Wang, G., Mehta, R., Badve, S., Thomson, M., Ham-
mond, S., Steeg, P., Liu, Y., and Nakshatri, H. (2011).
Control of EVI-1 oncogene expression in metastatic
breast cancer cells through microRNA mir-22. Onco-
gene, 30(11):1290–1301.
Patnala, R., Clements, J., and Batra, J. (2013). Candi-
date gene association studies: a comprehensive guide
to useful in silico tools. BMC Genet., 14:39:doi:
10.1186/1471–2156–14–39.
Rocchi, A., Manara, M., Sciandra, M., Zambelli, D., Nardi,
F., Nicoletti, G., Garofalo, C., Meschini, S., Astolfi,
A., Colombo, M., Lessnick, S., Picci, P., and Scot-
landi, K. (2010). CD99 inhibits neural differentiation
of human ewing sarcoma cells and thereby contributes
to oncogenesis. J Clin Invest, 120(3):668–680.
Slaby, O., Bienertova-Vasku, J., Svoboda, M., and Vyzula,
R. (2012). Genetic polymorphisms and microRNAs:
new direction in molecular epidemiology of solid can-
cer. J Cell Mol Med., 16(1):8–21.
Szkiba, D., Kapun, M., von Haeseler, A., and Gallach,
M. (2014). SNP2GO: functional analysis of genome-
wide association studies. Genetics., 197(1):285–289.
Vergoulis, T., Vlachos, T., Alexiou, P., Georgakilas, G.,
Maragkakis, M., Reczko, M., Gerangelos, S., Koziris,
N., Dalamagas, T., and Hatzigeorgiou, A. (2012). Tar-
base 6.0: Capturing the exponential growth of mirna
targets with experimental support. Nucleic Acids Re-
sources, 40(D1):222–229.
Vitale, A., Tan, H., and Jin, P. (2011). MicroRNAs, SNPSs
and cancer. Nucleic Acids Invest., 2(6):32–38.
Wang, K., Li, M., and Hakonarson, H. (2010). Analysing
biological pathways in genome-wide association stud-
ies. Nat. Rev. Genet., 11:843–854.
Xu, Z. and Taylor, J. (2009). SNPinfo: integrating GWAS
and candidate gene information into functional SNP
selection for genetic association studies. Nucl. Acids
Res., (Web Server issue):W600–5.
Yamada, A., Ishikawa, T., Ota, I., Kimura, M., Shimizu,
D., Tanabe, M., Chishima, T., Sasaki, T., Ichikawa,
Y., Morita, S., Yoshiura, K., Takabe, K., and Endo,
I. (2013). High expression of ATP-binding cassette
transporter ABCC11 in breast tumors is associated
with aggressive subtypes and low disease-free sur-
vival. Breast Cancer Res Treat., 137(3):773–782.
Yu, F., Yao, H., Zhu, P., Zhang, X., Pan, Q., Gong, C.,
Huang, Y., Hu, X., Su, F., Lieberman, J., and Song, E.
(2007). let-7 regulates self renewal and tumorigenicity
of breast cancer cells. Cell, 131(6):1109 – 1123.
Yu, J. and Zhang, L. (2005). The transcriptional targets of
p53 in apoptosis control. Biochem Biophys Res Com-
mun., 3(331):851–858.
Zhao, L., Bode, A., Cao, Y., and Dong, Z. (2012).
Regulatory mechanisms and clinical perspectives of
miRNA in tumor radiosensitivity. Carcinogenesis.,
33(11):2220–2227.
Zyla, J., Badie, C., Alsbeih, G., and Polanska, J. (2014).
Modelling of genetic interactions in GWAS reveals
more complex relations between genotype and phe-
notype. In Proceeding of: Bioinformatics 2014: 5th
International Conference on Bioinformatics Models,
Methods and Algorithms. SCITEPRESS.
BIOINFORMATICS2015-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
250
