
An Extension to Local Network Alignment using
Hidden Markov Models (HMMs)
Hakan G
¨
und
¨
uz
1
and
˙
Ibrahim S
¨
uzer
2
1
Department of Computer Engineering, Istanbul Technical University, Istanbul, Turkey
2
Department of Computer Engineering, Bo
˘
gazic¸i University, Istanbul, Turkey
Keywords:
Biological Networks, Local Network Alignment, Hidden Markov Models.
Abstract:
Local alignment is done on biological networks to find common conserved substructures belonging to different
organisms. Many algorithms such as PathBLAST (Kelley et al., 2003), Network-BLAST (Scott et al., 2006)
are used to align networks locally and they are generally good at finding small sized common substructures.
However, these algorithms have same failures about finding larger substructures because of complexity issues.
To overcome these issues, Hidden Markov Models (HMMs) is used. The study done by (Qian and Yoon, 2009),
uses HMMs to find optimal conserved paths in two biological networks where aligned paths have constant path
length. In this paper, we aim to make an extension to the local network alignment procedure done in (Qian
and Yoon, 2009) to find common substructures in varying length sizes between the biological networks. We
again used same algorithm to find k-length exact matches from networks and we used them to find common
substructures in two forms as sub-graphs and extended paths. These structures do not need to have the same
number of nodes and should satisfy the predefined similarity threshold (s0). The other parameter is the length
of exact paths (k) formed from biological networks and choosing a lower k value is faster but bigger values
might be needed in order to balance the number of matching paths below s0.
1 INTRODUCTION
With the advances in computer science and bioinfor-
matics, molecular and biological interactions are ex-
amined in a systematic way between different organ-
isms (Von Mering et al., 2002). In order to gain useful
information from organisms, graph structured biolog-
ical networks are used. In these networks, nodes rep-
resent the basic entities like proteins and the edges
between them show the interactions. When we have
biological networks of different organisms, we can
align them to compare and find the common substruc-
tures. This process is known as network alignment.
Network alignment also helps us find out the con-
served functional modules and their detailed molecu-
lar mechanisms form these functions. Network align-
ment is so important to detect the conserved interac-
tion patterns and many network alignment algorithms
are aimed to do this. The detected patterns in the
networks will be biological pathways that are known
or statistically significant pathways that are explored.
Network alignment is separated into two groups as
global alignment and local alignment. In global net-
work alignments, the aim is to find and optimize the
best overall match between two biological networks.
For this reason, the global network alignment prob-
lem can be thought as a graph matching problem. Lo-
cal network alignments try to find small subsets of
global ones and they basically aim to find common
substructures between two biological networks.
In this paper, we design an extension to lo-
cal alignment algorithm used in biological networks
belonging to different organisms based on Hidden
Markov Models (HMMs). We devise an algorithm
that finds the conserved substructures of varying sizes
instead of fixed lengths done in (Qian and Yoon,
2009). We detect two types of common substructures
from aligned networks with this algorithm.
Also, it is important to mention that our exten-
sion does not find optimal substructures but runs as
a heuristic algorithm using fixed length optimal paths
to construct bigger structures. We aim to use the idea
of obtaining smaller exact matches then trying to ex-
tend and combine them to build bigger but relevant
structures. This is similar to how BLAST type algo-
rithms work (Kelley et al., 2003) (Scott et al., 2006).
Our main intention is to overcome complexity issue
that arises when working on big networks while min-
Gündüz, H. and Süzer,
˙
I.
An Extension to Local Network Alignment using Hidden Markov Models (HMMs).
DOI: 10.5220/0005742102530257
In Proceedings of the 9th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2016) - Volume 3: BIOINFORMATICS, pages 253-257
ISBN: 978-989-758-170-0
Copyright
c
2016 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
253

imizing downside of not finding optimal solutions.
The paper continues as follows: In Section 2, we
summarize related work in the literature about net-
work alignments. In the 3rd section, we define our
methods and give some visual examples about them.
Section 4 gives information about the evaluations and
the strengths of the algorithm. In final section, we
conclude the paper.
2 RELATED WORK
There are many algorithms used on global and local
network alignment problems in the literature. Global
network alignment problems are solved via various
techniques such as integer programming (Li et al.,
2007), spectral clustering (Liao et al., 2009) and mes-
sage passing (Zaslavskiy et al., 2009). While lo-
cal network alignment problem is solved by differ-
ent kinds of algorithms and applications. PathBLAST
(Kelley et al., 2003), Network-BLAST (Scott et al.,
2006), QPath (Shlomi et al., 2006), PathMatch and
GraphMatch (Yang and Sze, 2007) are generally used
in finding conservative structures in biological net-
works. Most used algorithm in literature is Path-
BLAST which is an efficient algorithm for aligning
two Protein Protein Interaction (PPI) networks. This
algorithm looks for high-scoring pathway alignments
by considering the homology between aligned pro-
teins. When PPI data are noisy, it can allow gaps and
mismatches to handle variations (Kelley et al., 2003).
Local network alignment algorithms are generally
good at finding small sized common substructures in
given networks but they have same failures about find-
ing larger substructures because of complexity issues.
Also, some of these algorithms do not give chance to
node insertions and deletions in the alignment pro-
cess. In order to handle mentioned issues, Hidden
Markov Models (HMMs) based local network align-
ment is introduced in (Qian and Yoon, 2009). HMMs
has ability to combine node similarities and interac-
tion reliabilities (transition probabilities) to compare
aligned paths and they can also overcome the path iso-
morphism. In (Qian and Yoon, 2009), the researchers
adopt the HMMs framework to find optimal and bio-
logically significant paths in general biological net-
works. Their main goal is to find conserved paths
in two or more biological networks which have sim-
ilarities. They used a scoring scheme to find align-
ments and they search for top k alignments of homol-
ogous paths with the highest scores. Their extended
algorithms has polynomial complexity and it is de-
pendent on the length of aligned paths and the num-
ber of interactions (edges) between each networks.
Aligned paths may have insertions and/or deletions.
After finding high scoring paths, we will attempt to
combine overlapped ones to form the conserved sub-
networks in general network structure.
3 METHODS
In this section we will present an extension to the al-
gorithm described in (Qian and Yoon, 2009) for find-
ing the conserved substructures of varying sizes. The
algorithm presented in (Qian and Yoon, 2009) uses
HMMs for solving local network alignment problem
to find the optimal paths of fixed length.
The details how to do pairwise local alignment in
study (Qian and Yoon, 2009) is stated in 3.1.
3.1 Pairwise Local Alignment
We assume that we have two graphs as G1= (u,d)
and G2=(v,e) representing two biological networks.
The graph G1 has 10 nodes represented as u1, u2
. . . u10 respectively. Also, it has edges between the
nodes which shows the interaction between each en-
tity (node). The graph G2 has 9 nodes named from v1
to v9 and it has again the edges between the nodes
interacted each other. The example of two graphs
shown in Figure 1. These networks are undirected
that means there are same relations between node i to
node j and node j to node i.
For example, when G1 represents a PPI network,
each ui corresponds to a protein, and the edge be-
tween ui and uj shows that these proteins can interact
to each other. When we look at the interacting pair
nodes ie. (ui,uj), the interaction reliability is defined
as w1(ui, uj). It can be accepted as a weight of edge
between node ui and node uj (dij).Similarly, the in-
teraction reliability between two nodes vi and vj in
the graph G2 can be stated as w2(vi,vj).
Finally, the similarity between two nodes ui from
G1 and vj from G2 in the shown networks is defined
as h(ui,vj) and it is found by sequence similarity be-
tween two nodes The aim is to find the best matching
pair of paths from two networks maximizing defined
path alignment score that uses w1(ui, uj), w2(vi,vj),
h(ui,vj) and penalty gap scores. The main strength of
the algorithm is to find pairs of paths have fixed length
size.
Figure 2 shows an example of an alignment be-
tween two similar paths s and q, where s belongs to
G1 and q belongs to G2. The dashed lines in Figure
1 connect two nodes ui and vj indicate that there exist
significant similarities between the connected nodes.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
254
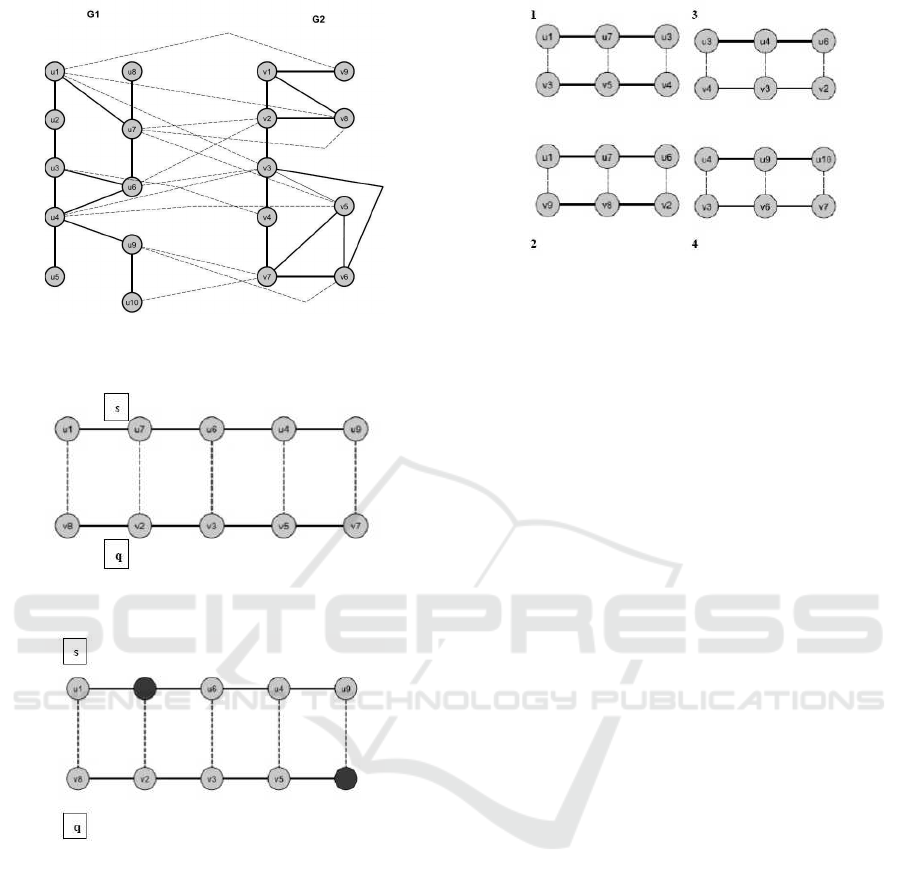
Figure 1: The example of two biological networks (G1 and
G2).
Figure 2: . An example of an ungapped alignment between
two similar paths s and q.
Figure 3: An example of a gapped alignment between two
similar paths s and q.
In the example shown in Figure 3, the optimal
alignment that maximizes the alignment score is taken
with gapped alignment. The aligned pair has two gaps
at v2 and u9. Insertion and deletion operations are
correlated to each other and an insertion in s (e.g. u9)
can be thought as a deletion in the aligned path q. In
the next subsection, we mention how we can extend
the algorithm used in (Qian and Yoon, 2009).
3.2 Extension
The extension we did contains mainly two steps:
Firstly, we will use (Qian and Yoon, 2009) to find
the best matching k-length path for pairwise local
alignment. Then, we will store this path and hide
Figure 4: Collection of distinct aligned paths from G1 and
G2 (k=3).
edges used by this path. The reason for hiding is to
not find and use same paths later. After this, we will
iteratively run this algorithm (Qian and Yoon, 2009)
to find the next matching k-length path that does not
contain any edges with previous best path. We will
continue this until we find all paths with intended
similarity threshold or of certain number threshold
(s0=determined threshold similarity). Finally, we will
have a collection of distinct aligned paths. For k=3,
the samples of distinct aligned paths got from G1 and
G2 are illustrated in Figure 4. Our extension will try
to combine these distinct paths into relevant types of
structures as subgraphs and longer paths. The details
of structures are explained in next subsections.
3.3 For Finding Subgraphs
To combine collected paths, we will find the paths that
contain same nodes and merge them. Since none of
them have any common edges, there will not be any
overlap. At the end of this procedure we will have the
number of distinct subgraphs of G1 and G2 shown
in Figure 5. This virtual subgraph does not need to
represent the same number of nodes in both graphs.
Because, all paths we are trying to combine are actu-
ally represented by a pair of nodes from G1 and G2
in each of its nodes. This leads to possibility of dif-
ferent nodes in one of the graphs can be mapped to
same node in other. The resulting subgraphs are all
conserved substructures that adhere to the similarity
threshold for their paths.
3.4 For Finding Longer Paths
Different from finding subgraphs, we will only try to
extend our structure as a path. To this end, we will
only combine our set of collected paths at their end
points. We cant just combine them just by their edges
like for subgraph case because just having distinct
edges do not allow us to combine paths from their
An Extension to Local Network Alignment using Hidden Markov Models (HMMs)
255
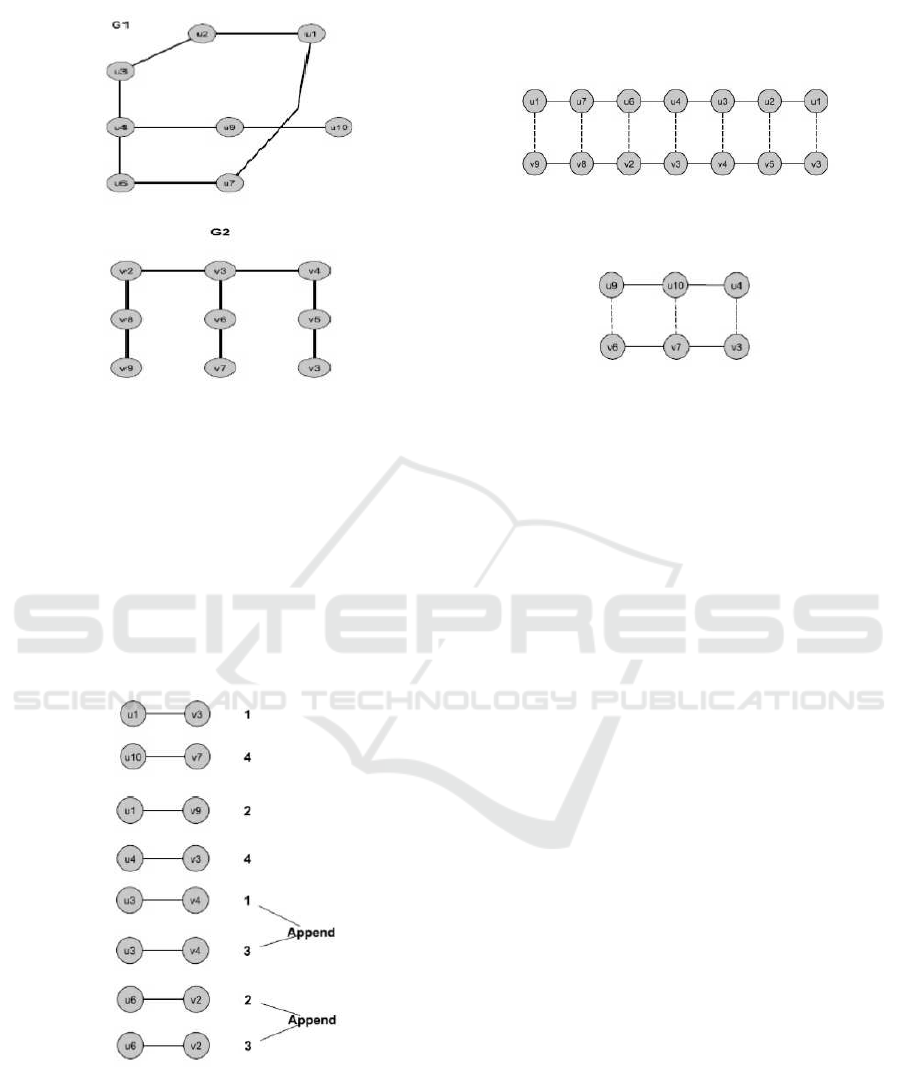
Figure 5: Distinct subgraphs of G1 and G2.
endpoints. To overcome this problem, we will con-
sider only endpoints and sort them by the number of
same endpoints. We do not need to consider isolated
end points. And endpoints with number of 2 can only
be appended to each other. So, we combine them and
only consider the choice with endpoints numbering 3
or more. For these, we will look at the length of can-
didate paths. Figure 6 shows the endpoints of distinct
aligned paths. The numbers at right of the each node
explains the corresponding paths (Figure 4).
Figure 6: Candidate paths endpoints.
Since we have already considered the similar-
ity scores for choosing paths with (Qian and Yoon,
2009), we will now try to get longer paths and choose
the longest two paths for appending and continue un-
til only one or no candidate remains. Since we do not
consider isolated endpoints, we can safely ignore this
endpoints. At the end of the procedure, we will have
a collection of paths with differing lengths.
Figure 7: Paths generated from candidate paths.
The distinct paths had the same endpoints are ap-
pended in order to generate longer paths. The exam-
ples of paths are shown in Figure 7.
4 EVALUATION
4.1 Possible Problems
Unlike the algorithm from (Qian and Yoon, 2009),
our solutions are not aimed to be optimal thus we use
heuristics approach. But we intend to reach near op-
timal solutions nevertheless.
• For path selection choosing by length may lead to
losing possible longer path. But considering all
pos-sible pairs for appending least to an exponen-
tial time complexity.
• For more than 2 graphs, our extension may not
work properly.
4.2 Parameters
When choosing parameters that effect the computa-
tion of algorithm and the quality of solutions, there is
a need to test which values are the best.
• k : length of matching pairs from (Qian and Yoon,
2009). Choosing a lower value is faster but big-
ger values might be needed in order to balance the
number of matching paths below s0
• s0 : Similarity score threshold for k-length match-
ing pairs. This effects number of matching pairs
we need to combine and run (Qian and Yoon,
2009). Thus, it directly decides computation time.
And value of k changes the values of scores even
for same network so s0 is also dependent on k.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
256

4.3 Test Cases
In order to check if structures we find are biologically
significant, we need to compare them with real-world
data. Similar to (Qian and Yoon, 2009), we will plan
to use data from KEGG (Kanehisa and Goto, 2000)
database in order to check if we can find the signifi-
cant structures.
In order to check the error rate of our algo-
rithm, we also need to try to compare same graphs.
Since checking isomorphism is an NP-Hard problem
(D
¨
opmann, 2013), we will undoubtedly wont be able
to give the exact graph but by checking how close
the answers are for same graph we can compare its
results with other graphs to understand how similar
structured we can expect.
5 CONCLUSIONS
By using the described technique, we would expect to
get significant similar substructures between the pair
of distinct biological networks. Different from the al-
gorithm we extend, we change problem into finding
big substructures within a similarity constraint. We
also consider two types of substructures and can ex-
tend it for different substructures. Sadly, our exten-
sion is not expected to get good results for multiple
network case. As a possible future work, multiple
alignment case can also be considered. Also as men-
tioned, this is a heuristics approach and there may be
a lot room to gain better results. But these improve-
ments require experimental results first.
REFERENCES
D
¨
opmann, C. (2013). Survey on the graph alignment prob-
lem and a benchmark of suitable algorithms.
Kanehisa, M. and Goto, S. (2000). Kegg: kyoto encyclo-
pedia of genes and genomes. Nucleic acids research,
28(1):27–30.
Kelley, B. P., Sharan, R., Karp, R. M., Sittler, T., Root,
D. E., Stockwell, B. R., and Ideker, T. (2003). Con-
served pathways within bacteria and yeast as revealed
by global protein network alignment. Proceedings of
the National Academy of Sciences, 100(20):11394–
11399.
Li, Z., Zhang, S., Wang, Y., Zhang, X.-S., and Chen,
L. (2007). Alignment of molecular networks by
integer quadratic programming. Bioinformatics,
23(13):1631–1639.
Liao, C.-S., Lu, K., Baym, M., Singh, R., and Berger, B.
(2009). Isorankn: spectral methods for global align-
ment of multiple protein networks. Bioinformatics,
25(12):i253–i258.
Qian, X. and Yoon, B.-J. (2009). Effective identification
of conserved pathways in biological networks using
hidden markov models. PLoS One, 4(12):e8070.
Scott, J., Ideker, T., Karp, R. M., and Sharan, R. (2006).
Efficient algorithms for detecting signaling pathways
in protein interaction networks. Journal of Computa-
tional Biology, 13(2):133–144.
Shlomi, T., Segal, D., Ruppin, E., and Sharan, R. (2006).
Qpath: a method for querying pathways in a protein-
protein interaction network. BMC bioinformatics,
7(1):199.
Von Mering, C., Krause, R., Snel, B., Cornell, M., Oliver,
S. G., Fields, S., and Bork, P. (2002). Comparative
assessment of large-scale data sets of protein–protein
interactions. Nature, 417(6887):399–403.
Yang, Q. and Sze, S.-H. (2007). Path matching and graph
matching in biological networks. Journal of Compu-
tational Biology, 14(1):56–67.
Zaslavskiy, M., Bach, F., and Vert, J.-P. (2009). Global
alignment of protein–protein interaction networks
by graph matching methods. Bioinformatics,
25(12):i259–1267.
APPENDIX
Pseudo Code of Algorithm
Set similarity threshold (s0)
Set length of exact matches (k)
Initialize similarity score (s) to s0
While s >= s0
-Find k-length exact match and similarity
score (s) using (Qian and Yoon, 2009)
-Trim edges of paths that emits the optimal
virtual path (hide).
if the desired structure is subgraph
-Iteratively find and merge in all net-
works that share same nodes in correspon-
ding networks
-Visualize subgraphs
else
-Iteratively find and append paths that
share same nodes in corresponding networks
at their endpoints
-Visualize extended paths
An Extension to Local Network Alignment using Hidden Markov Models (HMMs)
257
