
Two Novel Techniques for Space Compaction on Biological
Sequences
George Volis, Christos Makris and Andreas Kanavos
Department of Computer Engineering and Informatics, 26504 Rio, University of Patras, Patras, Greece
Keywords: Searching and Browsing, Web Information Filtering and Retrieval, Text Mining, Indexing Structures,
Inverted Files, Index Compression-Gram Indexing, Sequence Analysis and Assembly.
Abstract: The number and size of genomic databases have grown rapidly the last years. Consequently, the number of
Internet-accessible databases has been rapidly growing .Therefore there is a need for satisfactory methods for
managing this growing information. A lot of effort has been put to this direction. Contributing to this effort
this paper presents two algorithms which can eliminate the amount of space for storing genomic information.
Our first algorithm is based on the classic n-grams/2L technique for indexing a DNA sequence and it can
convert the Inverted Index of this classic algorithm to a more compressed format. Researchers have revealed
the existence of repeated and palindrome patterns in DNA of living organisms. The main motivation of this
technique is based on this remark and proposes an alternative data structure for handling these sequences. Our
experimental results show that our algorithm can achieve a more efficient index than the n-grams/2L algorithm
and can be adapted by any algorithm that is based to n-grams/2L The second algorithm is based on the n-
grams technique. Perceiving the four symbols of DNA alphabet as vertex of a square scheme imprint a DNA
sequence as a relation between vertices, sides and diagonals of a square. The experimental results shows that
this second idea succeed even more successfully compression of our index structure.
1 INTRODUCTION
The large volume of biological sequences demands
effective data structures and techniques for storing this
growing information. In addition, the DNA structure
analysis has shown that these sequences are not
random. This is somewhat expected if we consider that
DNA structure reflects the organizational structure of
living organisms so it must contain some logical
organization in its structure. One of the first things that
DNA sequencing disclosed was the occurrence of
repeated patterns in its body. It is well known
nowadays that the existence of repetitive sequences or
palindromes in a DNA sequence is one of the main
characteristics of DNA structure. We also know that
repeated DNA sequences are liable for biological
diversification (Grechko 2011) and that palindromic
sequences are associated with sites of DNA breakage
during gene conversion (Krawinke et al. 1986).
From that point a lot of techniques for the
identification of repeated or palindrome subsequences
came to the fore and lots of them proposed efficient
methods for handling sequences exploiting these
properties of the DNA structure. (Ziv and Lempel
1977), (Smith and Waterman 1981), (Welch 1984),
(Grumbach and Tahi 1994), (Rivals et al. 1995), (Kurtz
and Schleiermacher 1999), (Sun et al. 2004),
(Bernstein and Zobel 2004), (Christodoulakis et al.
2006), (Alatabbi et al.2012), (Diamanti et al. 2014).
The majority of them relies on the extraction of
repeated sequences. The detection of palindromes for
better performance in terms of space or time has
employed less research and that’s why palindrome
techniques lacked in literature in contrast to the
repeated sequence methods (Welch 1984), (Grumbach
and Tahi 1994), (Rivals et al. 1995).
The problem of a pattern detection into a DNA
sequence, a well-known problem of information
retrieval theory, is what we are going to solve. Our
main purpose is to achieve a better space performance.
Thus we propose two different techniques. The novelty
of our first approach relies on the combination of the
idea of palindromic sequences with n-grams to create
an alternative inverted index for our DNA sequence.
The second idea is a completely new technique which
is completely new technique which is based on
perceiving a DNA sequence as a geometric problem. It
is combined with the n-grams technique too.
Volis, G., Makris, C. and Kanavos, A.
Two Novel Techniques for Space Compaction on Biological Sequences.
In Proceedings of the 12th International Conference on Web Information Systems and Technologies (WEBIST 2016) - Volume 1, pages 105-112
ISBN: 978-989-758-186-1
Copyright
c
2016 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
105

In information retrieval theory the n-grams
technique seems very appealing for constructing the
index of a sequence (Navarro and Baeza-Yates 1998),
(Kim et al. 2005), (Mayfield and McNamee 2003).
This is due to the two major advantages of this
technique: a) its neutral language and b) its error
tolerance. Due to the first advantage we can disregard
the characteristics of the language and therefore we can
apply it at any language (Asian, Korean, and languages
where complicated knowledge is required) (Ogawa
and Iwasaki 1995), (Lee and Ahn 1996). The second
advantage allows us to retrieve information with some
errors (Kim et al. 2007).
The rest of the paper is organized as follows. In
section 2, the related work as well as the contribution
is presented. In section 3, we present the first
technique for Space compaction due to palindrome
extraction. Subsequently, in section 4, we present our
second technique and we analyze the square scheme
algorithm. The section 5 presents a reference to our
experimental results and the final section 6 concludes
the paper.
2 RELATED WORK AND
CONTRIBUTION
Finding the exact occurrences of a pattern in a
sequence of characters is one of the most fundamental
issues in information retrieval theory. Many types of
data structures and algorithms have been proposed
over the past years for effective solutions to this
problem. However it is still a challenging problem
when handling a big amount of data.
The n–grams technique (Navarro and Baeza-Yates
1998) in response to that problem is one of the well-
known and most used techniques over the past years.
We give the definition for n-grams below.
Definition n-grams: Given a sequence of tokens S
= (S₁, S₂……S
ɴ + (n-1)) over the token alphabet A,
where N and n are positive integers, an n-gram of the
sequence S is any n-long subsequence of consecutive
tokens. The i
th
-gram of S is the sequence (Sᵢ……Sᵢ +
(n-1)).
However, the n-grams structure for indexing a
sequence has some drawbacks too. The size of index
gets large and the performance of queries gets bad
too. That is a result of the 1-sliding method that n-
grams technique uses for extracting terms. It
increases the number of the extracted terms causing a
drastic increment of the size of the index. That also
affects the performance of queries since the number
of postings accessed during query performance
increases. For the reasons listed above a new data
structure was proposed. That was a two-level scheme
index that reduced the size and improved the query
performance (Kim et al. 2005).
The improved n-gram/2L algorithm that was
proposed for the reduction of index exponential
explosion is a two-level structure consisting from the
back-end and the front-end index. On the first level, the
algorithm extracts substrings of fixed length m from the
DNA sequence and stores them along with their offsets
to the back-end index. It subsequently applies the classic
n-grams technique for the set of extracted subsequences
and builds the front-end index.
Finding repeated sequences has been a basic step
for improving an information system performance
and reducing the amount of the requiring space. The
work of (Bernstein and Zobel 2004) proposed a
technique for computing repeated n-grams for large
text sequences. They proposed the SPEX multi pass
algorithm for finding co-occurring text. The LZ77
and LZ78 (Ziv and Lempel 1977) algorithms achieve
compression by replacing repeated pattern
occurrences in a sequence using references to a single
copy of that pattern, existing earlier in the
uncompressed data stream. Moreover, in (Diamanti et
al 2014) we can observe that taking advantage of the
repeatability of our subsequences in the back-end
index of the n-grams/2L scheme can produce a
smaller inverted index.
As we mentioned earlier, the repetitiousness of a
pattern can be revealed in terms of palindrome
existence. A palindrome of a pattern is the sequence
which arises if we traverse the pattern with reverse
order. For example, the AATTC pattern’s palindrome
is the CTTAA pattern.
The palindromes' extraction as a base for an
effective performance on genomics algorithms has
been investigated in (Grumbach and Tahi 1994) and
(Rivals et al. 1995). Both techniques are based on the
work of (Welch 1984) and achieves space
compression by searching for the longest exact and
Figure 1: Union of two palindrome grams.
AG m1,m3,m7,m8
GA m1,m3,m6,m8
AG m1,m3,m7,m8,m1,m3,m6,m8
AG m1,m3,m6,m7,m8
WEBIST 2016 - 12th International Conference on Web Information Systems and Technologies
106

reverse repetitions.
We should notice here that even if there is not an
appearance of repeated patterns palindromes as a
result of some structure mechanism of DNA, the fact
that the DNA alphabet is too small (only 4 characters)
leads us to the outcome that it is quite possible to find
palindromes or repeating patterns in a DNA data
stream.
Our first technique is concerned only to find
palindrome relations and take advantage of their
properties in order to achieve a better space
performance. We will also show that our idea could
be adapted from any algorithm that relies on the two
level scheme data structure for n-grams.
Bibliography lacks from references similar to our
second technique thus we will be the first to present
this approach for handling DNA sequences.
3 FIRST PROPOSED
TECHNIQUE (PALINDROME
ALGORITHM)
This novel two level scheme is based on the two-level
n-grams scheme of the work of (Kim et al. 2005). We
will separate the process in two steps. The first step
introduces the construction of our two-level index and
the second step applies our algorithm on the front-end
index.
STEP1
1: The initial DNA sequence produces the
subsequences of fixed m-length.
2: An inverted Index called back-end for the sub
sequences with the pointers of their initial index to the
DNA sequence for every subsequence.
3: All the n-grams in each substring are produced.
4: The inverted index called front-end index is
being created for all the grams of the subsequences
with the pointers to the subsequences that include the
n-grams.
STEP 2
At this step we apply our palindrome method to
the front-end index in order to attain a smaller index
for our DNA sequence.
The algorithm computes the palindrome relation
between the grams of our front-end index. At first we
check every gram of our index and scan our list to see
if it contains its palindrome. When we find a
palindrome of a gram we concatenate the two posting
lists, the list of the gram and the list of its palindrome.
What we need to remark at this point is that
searching for a palindrome of a pattern is exactly the
same as looking for the same pattern in the sequence.
The only difference is that when looking for an exact
match we scan the sequence from left to right until we
find the pattern, while in the second occasion when
we look for the palindrome we need to scan the
sequence from right to left until we find an exact
match for our given pattern.
Remark: It is not necessary to keep two separate
lists for two grams that are palindromes. We can
delete all the terms of a gram posting list and merge
it with the list of its palindrome gram. Figure 1 depicts
the adjustment of this idea on two palindrome grams.
Obviously we don’t have to keep the same indexes
for a gram so we can eliminate these same positions
from our posting lists as shown to figure 1. We can
already notice that our front-end index transformed
into a more compressed format based on the idea
above. Also, we can see that we are talking about a
lossless algorithm since no information gets lost
during this alteration. All the information that was
included in the list of the GA gram can be found in
the AG gram list now.
It is clear that since we have got a merged list for
two palindromes, our algorithm needs a refining step
for ensuring the validity of our results. Thus, every
time we search for a pattern in a sequence we ensure
that we check the right gram of a list and not its
palindrome.
The previous method can be directly adapted by
the two-level scheme structure and offer to the front-
end index a more compacted format. This is very
important if we consider that the front-end index is
responsible for the drastic increment of the size of the
two-level scheme data structure.
In conclusion, we observe that after this
transformation our new index occupies less space. It
is clear that if we had a bigger subsequence we could
possibly find more palindromes that could lead to an
even more compressed format of our table.
4 SECOND TECHNIQUE
(SQUARE SCHEME
REPRESENTATION)
The novelty of this idea relies on a different approach
of the way we encounter a DNA sequence. Instead of
considering a DNA sequence as a random sequence,
which consists of symbols of a given DNA alphabet,
we consider it as a depiction of the relation of a
square’s sides, diagonal and vertices.
In that way genomic problems could acquire
geometrical concept. The assignment of DNA
characters to square vertexes is obviously a one-to-
Two Novel Techniques for Space Compaction on Biological Sequences
107

one correspondence, which indicates that there is no
loss of information after the substitution. The above
mentioned idea is illustrated on Figure 2
Figure 2: DNA bases as square vertices.
Hence, a DNA sequence can be transformed into
a vertex traversing depiction of a square. Assuming
that every two consecutive symbols in DNA sequence
are not identical we can transform the vertex
traversing into a sides and diagonals traversing
problem.
Following the above approach in the sequence S
=ACGT we may observe the depiction of two parallel
sides of the square which are the {AC} and the {GT}
sides. This is the way that the concept of the idea
works.
4.1 Square Scheme
At this stage we use a definition from mathematics
field in order to proceed with our technique. The
concept of the absolute value is introduced. For
example for a two character string S=GA, we call
absolute value of S, and we symbolize, it as |S| or
|GA|, the sequence that arise if the first character is
lexicographically smaller than the second character.
So |GA| =AG.
We will convert our DNA sequence to a new one
based on the square scheme and the absolute value
concept. We scan our sequence, extracting all the
consecutive 2 –character strings that do not overlap
each other. Every extracted string is converted to its
absolute value. For example we are indifferent about
the succession of characters in every extracted string.
Either it is AG or GA, we will consider it as AG.
Based on the above remarks we will take this
analysis on a further step. First of all we segment our
DNA sequence in corpuses of four DNA characters that
do not overlap each other. Every corpus can be divided
in two pairs of 2-character strings that do not overlap
each other and (if every two symbols are not identical)
can be transformed into a depiction of square sides and
diagonals, provided that in every pair of strings the
characters are not identical. (We will confront the case
of identical characters later).
Considering that every corpus of four DNA
symbols depicts a relation of square diagonals and
sides we can divide every DNA sequence to certain
categories based on these relations. Thus if we find
all the possible associations between square sides and
diagonals we can correspond every four DNA
characters string to a certain association of this square
scheme.
We now check all the relations that occur between
two square elements:
1) Parallel Sides: The four DNA symbols encode
two sides that are parallel. That occurs at the
following four cases:
Α) ΑC – GT B) GT - AC
C) AG – CT D) CT - AG
As we mentioned before we transpose the two 2-
character strings that compose the 4 symbol string
that we examine, into their absolute values.
2) Vertical Sides: We have vertical sides on the
following cases:
Α) AC – AG Ε) GT - CT
Β) AG – AC F) CT - GT
C) AG – GT G) CT - AT
D) GT – AG H) AT - CT
So we have finally 8 different categories.
3) A Side with a Diagonal: Each side can be
paired with the two diagonals so we have eight
categories related to this case:
A) AC – AT E) CT - AT
B) AC – CG F) CT - CG
C) AG – AT G) GT - AT
D) AG – CG H) GT - CG
If we consider the reverse relations, where the first
pair forms a diagonal and the second pair forms a side
of the square, we have finally sixteen categories.
4) Diagonal with Diagonal: This occurs on the
following four cases.
Α) AT – CG C) AT - AT
Β) CG – AT D) CG| - CG
5) Repeated Sides: This occurs on the following
four cases:
Α) AC - AC C) GT - GT
B) AG – AG D) CT - CT
Figure 3 illustrates the process of the proposed
technique.
All the above categories are related to the case in
which, the 4-character DNA corpus is divided in two
character string pairs, while the characters are not
identical. From a geometrical aspect, all the above
cases, can be expressed as a traversal on a square.
In the case that a repeated character appears in an
extracted 2-character string, indicates that we don’t
observe any movement in the square scheme thus no
side or diagonal is forming. In this case we assume
that we are indifferent on which is the repeated
WEBIST 2016 - 12th International Conference on Web Information Systems and Technologies
108

Figure 3: At S1 sequence we have substituted the 2 –
character consecutive strings that consist the S1 with their
absolute values. At S2 we segment our new sequence in
corpuses of four characters in order to encode them with
their geometrical correlation. At S3 we can see our new
sequence after the encoding.
character (vertex) appearing in each 2-character
string. For instance, in case we trace GG string (same
for CC or TT), we have to classify the string on the
same category that we would in case we had traced
the AA vertex. At a later stage, we will clarify, which
character appears in each 2-character string pair that
divides every 4-character DNA corpus. Thus we will
group together the following two categories on the
above case analysis
6) Two Repeated Vertexes: No movement at all.
(Repeated characters at both 2-character pair strings):
Since every repeated vertex is encountered as AA we
have only one case reflecting to this category.
A) AA - AA
7) One Repeated Vertex: The repeated vertex
can be paired with the two diagonals plus the four
vertices which means that we have totally six possible
relations in this case.
A) AA – AC D) AA - GT
B) AA – AG E) AA - AT
C) AA – CG F) AA - CG
Considering the reverse relations where the first
pair forms a diagonal or a side and the second pair
forms the repeated vertex (AA) we finally get twelve
cases.
4.2 Converting the Sequence to a New
One based to Square Scheme
Categories
Thus if we traverse the DNA sequence and encode
every corpus of four characters with the above
technique we can produce a new sequence where each
corpus is a representative of the category it belongs
in.
Encoding each category of the above scheme, in a
single symbol we can produce a new sequence where
every 4 symbols string corresponds to a new single
symbol. All the possible 4-character strings can be
classified to the 49 categories we have previously
described. Consequently, using the English alphabet
(Lowercase and Uppercase) and a terminal symbol
(#) for the case of repeated Vertex (AAAA) we will
be able to represent all these cases using just one
symbol for each one of these. Using this encoding, we
can reduce the size N of our initial sequence to a size
of N/4.We will save the index of the new sequence
using the one –level n-gram scheme and not the two-
level n-grams/2L .This is due to the structure of our
new alphabet. The sequence, contains a large number
of different symbols and that implies that we have less
possibilities to encounter repeated subsequences.
Thus, based on the relevant literature (Diamanti et al.
2014) we choose to save it on one-level scheme.
4.3 Converting the Pattern to a New
One based to Square Scheme
Category
At the next step we are going to encode our pattern to
our new alphabet in order to apply the n-grams
technique. Following the same approach we can
convert our initial pattern of size P to a new one of
size P/4.
After we have encoded our pattern, due to the
square scheme, we can apply the well-known n-grams
technique to extract its occurrences in the
DNA
sequence. But our process won’t stop there. There are
more to be done in order to extract all the appearances
of our initial pattern. Let’s give a simple example to
clarify this:
Assuming we have the pattern P =AGCTATGA
which will be segmented to the following strings:
AGCT - ATGA
1st 2nd
The pattern P will be encoded as P΄= {Parallel
sides A} {Side with diagonal B}. The problem here
is that we only search for encoded strings of the initial
pattern that have been encoded with this specific
order. But what happens if the AG has not been
encoded as the first two characters of a 4-character
string but it has been encoded as the last two
characters of another 4-character string? For example,
the pattern M =AGAGCTATGACT which with the
above technique will be segmented to the following
three strings:
S=ACTGATGCACACAGAC
S1=ACGTATCGACACAGAC
S2=ACGT ATCG ACAC AGAC
S3= {Parallel sides A}{Diagonal with diagonal
A}{Repeated side A}{Vertical sides B}
Two Novel Techniques for Space Compaction on Biological Sequences
109
AGAG - CTAT - GACT
1
st 2nd 3rd
The previous pattern will be encoded as M΄=
{repeated side B} {side with a diagonal D} {Parallel
side C}. So even if the pattern AGCTATGA is still
there, we cannot find it because we will be looking
for a different sequence of symbols after the encoding
(P΄ # M΄). This remark leads us to the conclusion that
in order to trace a pattern of DNA sequence after the
encoding we have to look not only for one pattern .In
particular we have to look for four patterns. Every
pattern will start the encoding at each one of the four
starting positions of the pattern.
The remaining symbols that cannot form a 4-
character string cannot be encoded. Therefore we
check for the encoded corpus of the sequence first and
if we have a match we subsequently check if the
remaining DNA symbols are identical. We can see for
the example above that P3 pattern can now detect the
existence of our initial pattern in the M sequence.
Thus, instead of searching for a pattern of size P
in a sequence with N size we are looking for four
patterns of size P/4 each one into a sequence of total
size N/4.
4.4 Refining Results
After the occurrences of these patterns have been
traced we proceed with the final step of our method.
At this step the final results for the occurrences of our
pattern are derived. The occurrences which are
extracted until now are just a sign of possible
existence of our pattern in the current positions. That
is due to the simplifications we made on the first steps
of our algorithm. Specifically, this is due to the
following two factors.
1) We have estimated the absolute values of
every 2-character string which have been extracted
from our initial DNA sequence. For instance there is
Figure 4: Refining List for the Subsequence S.
no distinction if the AG or the GA string appears.
2) We have represented every repeated vertex
(character) by a single representation (the AA string).
For instance there is no distinction if it is the CC or
the GG string.
Hence, we need to refine the prospective results to
produce our final answers. To do this we maintain for
our initial DNA sequence (of size N) a list that is
called Refining List. We examine every pair of two
character string that compose each 4-character string
that have been extracted provided that these two
character strings do not overlap each other. If the first
character of each pair is lexicographically smaller
than the second we register “0” in our Refining List.
On the opposite occasion we register “1”. Obviously
the size of our Refining List will be N/2. For the
repeated characters of a two character string we made
the following convention for our Refining List:
• If we find AA we entry 0.
• If we find CC we entry 1.
• If we find GG we entry 2.
• If we find TT we entry 3.
Figure 4 exhibits the construction of our Refining
List for a given sequence.
This is all that is needed in order to detect the exact
DNA symbol at every position of our transformed
sequence. Assuming that in a certain position in our
DNA sequence, the pattern ACGT is traced.
According to the square scheme algorithm analysis
we ascertain that this pattern corresponds to the
“parallel sides A” category, supposing that this
category has been encoded with the symbol U.
Moreover, we trace in our Refining List the numbers
0 and 1 at the corresponding position. This implies
that we have the string ACTG in the current position
on the DNA sequence. With the above process we can
clarify which DNA symbol lies behind the encoded
string.
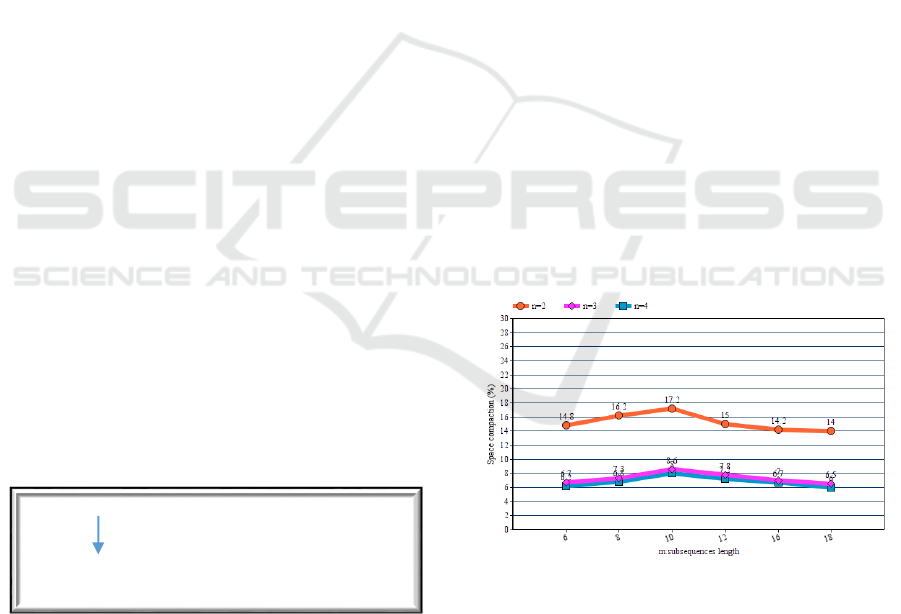
Figure 5: Percentage space compaction of palindrome
algorithm in comparison to n-grams/2L algorithm for
varying size of n.
After we have extracted the prospective sequences
that match our pattern, this technique is applied on
our pattern extraction step. At each one of our
equivalent patterns we also create a Refining List.
When there is a match at a current position we cross
examine the Refining List of our pattern with the
corpus of our DNA sequence Refining List that
S=ACGGTAGCAT
Refining List = 02110
WEBIST 2016 - 12th International Conference on Web Information Systems and Technologies
110
corresponds to the appearance of the pattern in the
sequence. If we have a match, then our pattern
definitely appears in our initial DNA sequence at this
current position. Otherwise there is no appearance of
this pattern at this position.
5 EXPERIMENTS
In our experiments we use random synthetic
sequences of 100000 DNA symbols in order to
examine the performance of our constructions and the
space compaction that they achieve. The computer
system, where the experiments were performed, was
an Intel Core i3-M380 2.53 GHz CPU with a 4GB
RAM. We used initials to describe the metrics of our
problem.
In particular we symbolize with m the length of
our subsequences and with n the length of grams. The
presentation of the results of each algorithm are
depicted in relation with the percentage of the space
compaction they achieve compared to the n-gram/2L
algorithm. In the first algorithm we estimated the
space efficiency of the front-end index (because the
method offers space efficiency only on the front-end)
while in the second algorithm we compared the whole
two-level index of the two techniques.
5.1 Palindrome Algorithm Results
As is depicted in figure 5 this method behaves better
and offers better space efficiency for the case of 2-
grams. This is a bit of expected if we consider that the
algorithm achieves compaction in case it detects
palindrome grams within the same subsequence.
There are more possibilities to find a palindrome of a
2-gram than palindromes of larger grams within a
subsequence. Since this technique exploits the
reciprocity of the DNA structure it is reasonable to
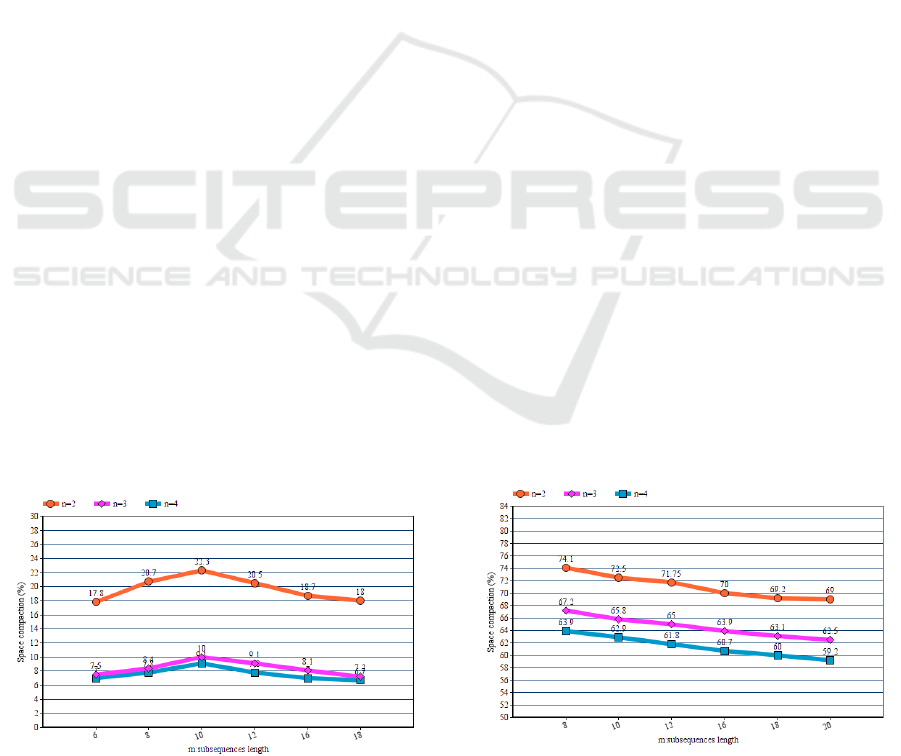
Figure 6: Percentage space compaction of palindrome
algorithm for palindromic DNA sequence in comparison to
n-grams/2L algorithm for varying size of n.
achieve even better efficiency on sequences which
show large volume of palindromes on their body. This
can be depicted in figure 6 where a higher space
compaction can be observed. In both figures we can
patently see that our method is more efficient for
substrings of length from 8 to 12.The reason for this
efficiency is that our palindrome algorithm takes
advantage of the substrings of length from 8 to 12
which seems to show larger volume of palindromes
on their body.
5.2 Square Scheme Algorithm Results
As far as the second technique is concerned, we can
observe that we are led to very compact inverted file
sizes (Figure 7). It is also very important to notice that
this method cannot be affected from the nature of
DNA structure (repetitions, palindromes).It is clear
from the algorithm analysis that this technique can
guarantee this high efficiency for every biological
sequence. Since this method uses the one level
scheme it doesn’t affected by the length of our
subsequences. That’s why we observe a slight
reduction of the efficiency for larger values of m. This
is due to a possible space compaction that appears to
the two level n-gram/2L technique in contrast to the
one –level n-gram for higher values of m.(Due to the
work of (Diamanti et al. 2014)this can happen
because there is a large number of repeated substring
on our back-end index).
6 GENERAL CONCLUSION
We presented two novel techniques that can lead us
to new compact inverted index file sizes. Especially
the palindrome algorithm can be perceived as a
“black box” and thus can be adapted by any algorithm
that uses the n-gram/2L technique in order to provide
Figure 7: Percentage space compaction of square scheme
algorithm in comparison to n-grams/2L algorithm for
varying size of n.
Two Novel Techniques for Space Compaction on Biological Sequences
111

a more compressed index. The second technique can
obviously offer higher efficiency especially when
handling a big amount of data. Moreover this new
approach for handling DNA sequences as a
geometrical problem could possibly lead in future to
new and efficient ideas about DNA algorithms.
REFERENCES
Alatabbi, A., Crochemore, M., Iliopoulos, C. S., and
Okanlawon, T. A. (2012). Overlapping repetitions in
weighted sequence. In International Information
Technology Conference (CUBE), pp. 435-440.
Bernstein, Y., & Zobel, J. (2004, January). A scalable
system for identifying co-derivative documents.
In String Processing and Information Retrieval (pp. 55-
67). Springer Berlin Heidelberg.
Christodoulakis, M., Iliopoulos, C. S., Mouchard,
L.,Perdikuri, K., Tsakalidis, A. K., and Tsichlas,
K.(2006). Computation of repetitions and regularities
of biologically weighted sequences. In Journal of
Computational Biology (JCB), Volume 13, pp. 1214-
1231.
Diamanti, K., Kanavos, A., Makris, C., & Tokis, T.(2014)
Handling Weighted Sequences Employing Inverted
Files and Suffix Trees,
Grechko, V. V. (2011). Repeated DNA sequences as an
engine of biological diversification. Molecular
Biology, 45(5), 704-727.
Grumbach, S. and Tahi, F., A new challenge for
compression algorithms: genetic sequences, J.
Information Processing and Management, 30(6):875-
866, 1994.
Kim, M.-S., Whang, K.-Y., and Lee, J.-G. (2007).
ngram/2l-approximation: a two-level n-gram inverted
index structure for approximate string matching. In
Computer Systems: Science and Engineering, Volume
22, Number 6.
Kim, M.-S., Whang, K.-Y., Lee, J.-G., and Lee, M.-J.
(2005). n-gram/2l: A space and time efficient twolevel.
n-gram inverted index structure. In International.
Conference on Very Large Databases (VLDB),
pp. 325-336.
Krawinkel, U., Zoebelein, G., & Bothwell, A. L. M. (1986).
Palindromic sequences are associated with sites of
DNA breakage during gene conversion.Nucleic acids
research, 14(9), 3871-3882.
Kurtz, S., & Schleiermacher, C. (1999). REPuter: fast
computation of maximal repeats in complete genomes.
Bioinformatics, 15(5), 426-427.
Lee, J. H. and Ahn, J. S. (1996). Using n-grams for korean.
text retrieval. In ACM SIGIR, pp. 216-224.
Mayfield, J. and McNamee, P. (2003). Single n-gram
stemming.In ACM SIGIR, pp. 415-416.
Millar, E., Shen, D., Liu, J., & Nicholas, C. (2006).
Performance and scalability of a large-scale n-gram
based information retrieval system. Journal of digital
information, 1(5).
Navarro, G., & Baeza-Yates, R. (1998). A practical q-gram
index for text retrieval allowing errors. CLEI Electronic
Journal, 1(2), 1.
Ogawa, Y. and Iwasaki, M. (1995). A new characterbased
indexing organization using frequency data for
japanese documents. In ACM SIGIR, pp. 121-129.
Rivals, E., Delahaye, J.-P., Dauchet, M., and Delgrange, O.,
A Guaranteed Compression Scheme for ´ Repetitive
DNA Sequences, LIFL Lille I University, technical
report IT-285, 1995.
Smith, T. F., & Waterman, M. S. (1981). Identification of
common molecular subsequences. Journal of
molecular biology
, 147(1), 195-197.
Sun, Z., Yang, J., and Deogun, J. S. (2004). Misae: A new
approach for regulatory motif extraction. In
Computational Systems Bioinformatics Conference
(CSB), pp.173-181.
Welch, T. A. (1984). A technique for high-performance
data compression computer, 6(17), 8-19..
Ziv, J., & Lempel, A. (1977). A universal algorithm for
sequential data compression. IEEE Transactions on
information theory, 23(3), 337-343.
WEBIST 2016 - 12th International Conference on Web Information Systems and Technologies
112
