
Molecular Dynamics Use in Personalized Cancer Medicine
Example of MET Y501C Mutation
Igor F. Tsigelny
1,2,3
, Razelle Kurzrock
1
,
Åge Aleksander Skjevik
2,4
,
Valentina L. Kouznetsova
1,2
and Sadakatsu Ikeda
1
1
Moores Cancer Center, Univesity of California at San Diego, La Jolla, California, U.S.A.
2
San Diego Supercomputer Center, Univesity of California at San Diego, La Jolla, California, U.S.A.
3
Department of Neurosciences, Univesity of California at San Diego, La Jolla, California, U.S.A.
4
Department of Biomedicine, University of Bergen, Bergen, Norway
Keywords: Personalized Cancer Medicine, Molecular Dynamics, Structure of Proteins, Sema Domain, MET, c-Met.
Abstract: We explored a possible new method for prediction of activating mutations in cancer-related proteins. This
method is based on elucidation of flexibility of proteins associated in activating complexes. Based on the
theory of intermediate binding complexes, the binding process is not only related to the three-dimensional
structure of proteins, but also to the four-dimensional set of possible conformations allowed by the flexible
regions of the involved members of the associated complex. Using molecular dynamics simulations, we found
that an Y501C mutation in the MET gene might activate it. Using this information, a specific drug that
functioned as a potent MET inhibitor was prescribed and had a salutary impact on the tumor.
1 INTRODUCTION
Elucidation of possible changes in protein activity
based on an aberration in its sequence is one of the
most important tasks of personalized medicine
(Tsigelny et al., 2015). One of the most powerful
databases in cancer medicine is Cosmic (Forbes et al.,
2011). Even if one would extract only the 50 most
frequent mutations in cancers in 600 proteins (which
are used most frequently for cancer diagnostics), it
would give 30000 aberrations. In some cases, such
aberrations are described in the literature and related
databases. Nevertheless, more than 90 percent of
them are not covered. Here, we address the problem
of how to elucidate possible activity changes that
occur due to these mutations. Replacement of a
residue in a pdb file may help if we have a simple case
when the aberration effect is obvious: change of a
charged residue to an oppositely charged residue in a
salt bridge, or insertion of a hydrophobic residue
instead of a hydrophilic residue that participates in
hydrogen bonding etc. At the same time, the effect of
a residue substitution might not be that obvious in
other cases. Here molecular dynamics (MD)
simulation might help elucidate the changes in the
ensemble of conformers that could affect the activity
of the protein or protein complex.
2 MET STRUCTURE AND
FUNCTION
The tyrosine kinase MET is a receptor for the ligand–
hepatocyte growth factor (HGF). It is known to be
involved in cancerogenesis. When activated (in many
cases because of a single amino acid replacement
mutation), it affects cells in a number of organs
creating invasive cancers (Stamos et al., 2004,
Montesano et al, 1991). The MET receptor has
significant structural similarity to Ron and Sea
receptors (Ronsin et al., 1993; Huff et al., 1993,
Stamos et al., 2004).
In order to be activated, MET requires binding of
its ligand—hepatocyte growth factor (HGF) that is
active only after proteolytic conversion to a two chain
configuration (Stamos et al., 2004; Hartmann et al.,
1992). Direct binding sites show that HGF-beta
chains bind to the extracellular domain of MET with
a K
d
of about 90 nM. Accordingly, binding of HGF is
crucial to activation of MET (Figure 1). Analysis of
the MET–HGF interface shows a set of moderately
complementary side chains on both sides.
Tsigelny, I., Kurzrock, R., Skjevik, Å., Kouznetsova, V. and Ikeda, S.
Molecular Dynamics Use in Personalized Cancer Medicine - Example of MET Y501C Mutation.
DOI: 10.5220/0005959500710074
In Proceedings of the 6th International Conference on Simulation and Modeling Methodologies, Technologies and Applications (SIMULTECH 2016), pages 71-74
ISBN: 978-989-758-199-1
Copyright
c
2016 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
71
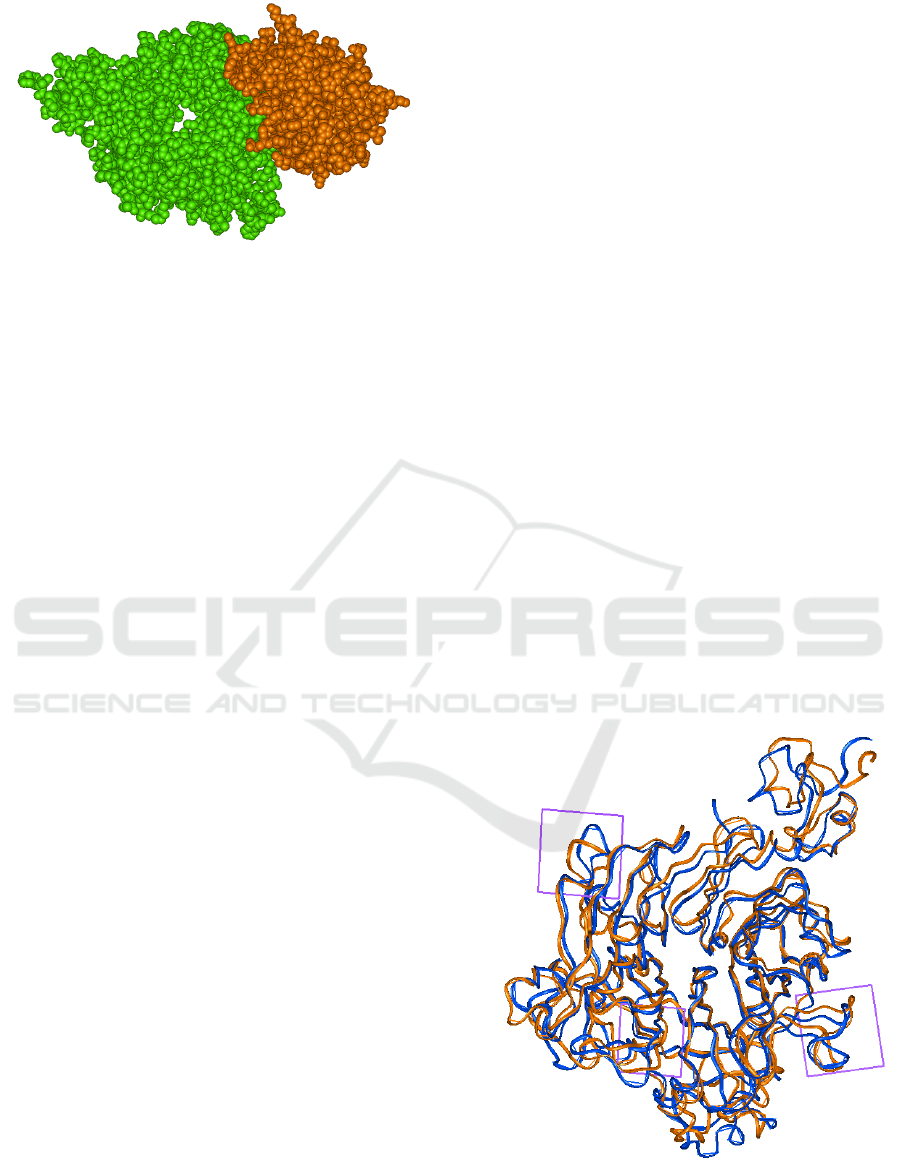
Figure 1: Interaction of the extracellular Sema domain of
Met (green) with the HGF-beta chain (brown).
2.1 Sema Domain
The Sema domain of MET forms a conformation
sometimes referred to as “seven-bladed beta-
propeller” in the shape of a funnel with an inner
diameter of 25 Å and a total diameter of around 50 Å.
The blades of this propeller are formed by antiparallel
beta-strands. The Sema domain is stabilized by the
interactions between the C- and N-terminal residues,
and the beta-propeller structure is stabilized by the
seven disulphide bridges also found in a number of
proteins with notable homology in the amino acids
included in these domains. The HGF beta-chain
associates with the Sema domain at the bottom face
of the propeller with at least seven electrostatic pas
interactions between the two proteins (Stamos et al.,
2004).
In the studied case, there is a mutation Y501C in
the Sema domain of MET. This residue is located at
the interface between the C- and N-terminal of the
Sema domain. From the general point of view used in
elucidation of possible activity changes of the MET–
HGF complex, a substitution of tyrosine to cysteine
would not make any changes in activity unless
cysteine is involved in a disulphide bond (not in this
case). Another possibility is that this mutation
happens in the N-C-terminal interface of the Sema
domain and affects its flexibility.
3 FLEXIBILITY OF BINDING
PROTEINS IMPROVES THEIR
ASSOCIATION
As we pointed above, the activation of the MET–HGF
complex significantly depends on interaction
between these two proteins. As was shown by Levi
and colleagues (2005), who studied more than 100
protein–protein complexes, the flexibility of the
binding partners is one very important feature that
often defines the process of protein–protein
association. In other words, the binding process is not
only related to the three-dimensional structure of
proteins, but also to the four-dimensional set of
possible conformations adopted by means of the
flexible regions of the involved proteins. It is
interesting to note that so-called transition-state
conformational ensembles for general folding of
proteins and their binding have similar characteristics
(Levi et al., 2005). Taking into consideration the
abovementioned concept, we hypothesized that
increasing flexibility of the Sema domain of MET
would increase its interactions with HGF and
consequently improve binding between these proteins
and thereby increase activation of the entire complex.
4 MUTATION Y501C AFFECTS
THE FLEXIBILITY OF THE
SEMA DOMAIN
In order to elucidate possible changes in flexibility of
the Sema domain, we conducted 300 ns MD for the
wild-type and mutated versions of the protein. Our
results show significant increase in flexibility of
several important parts of the Sema domain (Figure
2). The violet rectangles encompass the most flexible
regions of the mutant structure. It is interesting to note
that the maximum flexibility changes occur in the
Figure 2: Superposition of the 300 ns conformers, with the
wild-type represented by brown ribbons of the alpha-trace
and the Y501C mutant of the Sema domain of MET shown
as blue ribbons.
SIMULTECH 2016 - 6th International Conference on Simulation and Modeling Methodologies, Technologies and Applications
72
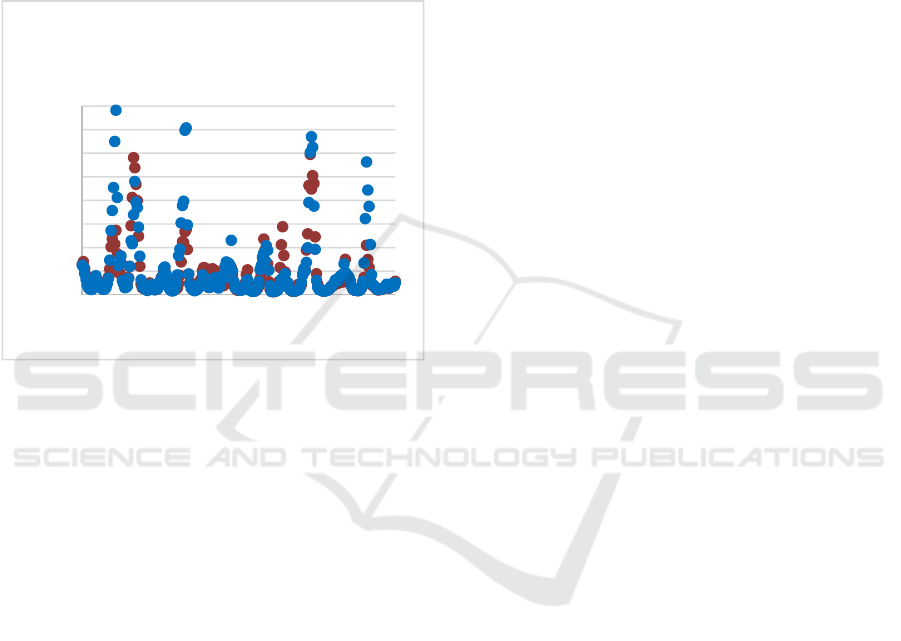
regions of the Sema domain that are in direct contact
with the HGF protein when bound (Figure 3). Note
that the regions around residues 150 and 210 of the
Sema domain are in direct contact with HGF residues
during its binding. These results suggest that the
mutation Y501C of MET leads to a significant
increase in the flexibility of the MET Sema domain
in the regions contacting HFG. Such changes may
improve the binding and consequently the activity of
the MET–HGF complex.
Figure 3: Flexibility (defined by B-factor values) of the
Sema domain residues in the wild-type (brown) and Y501C
mutant (blue) conformers as calculated from a 300 ns MD
trajectory.
5 PATIENT TREATMENT BASED
ON THE MD SIMULATIONS
A patient was diagnosed with hepatocellular
carcinoma (HCC), with the MET Y501C (tyrosine to
cysteine) missense mutation that was elucidated from
circulating tumor DNA. Based on the results of MD
simulation, we suggest that this mutation activates
MET kinase and consequently has oncogenic effects.
The patient received cabozantinib—a MET inhibitor.
This drug administration caused significant (65%)
reduction of alphapheto protein (AFP), a tumor
marker for HCC.
6 CONCLUSIONS
Molecular dynamics simulations can be used to
elucidate the four-dimensional ensembles of possible
conformers and flexibility of the binding partners in
protein–protein complexes and can help in decision
making for physicians in cancer therapy.
7 METHODS
In order to generate the Y462C Sema domain mutant,
Tyr462 in the wild-type protein was replaced by a
cysteine while the rest of the protein structure
remained unchanged. Disulfide bridges between the
relevant cysteine pairs in each protein were
generated. The SEMA domain of MET protein has
been extracted from the complex with heparin
(Stamos et al., 2004) pdb ID 1shy.
We conducted molecular dynamics (MD)
simulations for both the wild-type and mutated
versions of Sema domain of MET. The two proteins
were each placed in an octahedral water box
consisting of about 48,500 TIP3P water molecules
and 9 neutralizing K
+
ions modelled by
Joung/Cheatham ion parameters (Joung and
Cheatham, 2008). The simulations were conducted
using the GPU/CUDA-accelerated version of
PMEMD implemented in the AMBER14 software
suite (Case et al, 2014; Goetz et al., 2012; Salomon-
Ferrer et al., 2013A, B), with the protein described by
AMBER ff14 SB parameters. Each of the two protein
systems were subjected to the following
minimization/simulation steps: i) Unrestrained
minimization for 10,000 steps; ii) Gradual constant
volume heating from 0 to 100 K over 5 ps with
restraints applied to the protein backbone; iii) Gradual
constant pressure heating to 310 K over 100 ps with
restraints applied to the protein backbone; iv) 300 ns
unrestrained constant pressure simulation at 310 K.
The Langevin thermostat (Loncharich et al.,
1992) was applied for regulation of temperature with
a 1.0 ps
-1
collision frequency, and the pressure was
regulated isotropically during the second heating step
and the production simulation by means of the
Berendsen barostat (Berendsen et al., 1984) at a
reference pressure of 1.0 bar. Bond lengths for bonds
involving hydrogen were constrained using the
SHAKE algorithm (Rycjaert et al., 1984), allowing
for a time step of 2 fs. Periodic boundary conditions
were applied, and the particle mesh Ewald (PME)
method (Roe et al., 2013) was used for the evaluation
of electrostatics.
REFERENCES
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F.,
DiNola, A., Haak, J. R., 1984. Molecular dynamics
0
50
100
150
200
250
300
350
400
125 225 325
B‐factor
Residuenumber
Flexibilityoftheintialand
mutatedSemadomains
Molecular Dynamics Use in Personalized Cancer Medicine - Example of MET Y501C Mutation
73

with coupling to an external bath J. Chem. Phys., 1984,
81: 3684–3690.
Case, D. A., Babin, V., Berriman, J. T., et al., 2014. AMBER
14, University of California, San Francisco.
Darden, T., York, D., Pedersen, L., 1993. Particle mesh
Ewald: an Nlog(N) method for Ewald sums in large
systems, J. Chem. Phys., 98: 10089–10092.
Forbes, S. A., Bindal, N., Bamford, S., Cole, C., Kok Chai
Yin, Beare, D., Jia, M., Shepherd, R., Leung, K.,
Menzies, A., Teague, J. W., Campbell, P. J., Stratton,
M. R., Futreal, P. A., 2011. COSMIC: mining complete
cancer genomes in the Catalogue of Somatic Mutations
in Cancer, Nucleic Acids Res. (England), 39 (Database
issue): D945–D9450.
Götz, A. W., Williamson, M. J., Xu, D., Poole, D., Le
Grand, S., Walker, R. C., 2012. Routine microsecond
molecular dynamics simulations with AMBER on
GPUs. 1. Generalized Born, J. Chem. Theory Comput.,
8: 1542–1555.
Hartmann, G., Naldini, L., Weidner, K. M., Sachs, M.,
Vigna, E., Comoglio, P. M., Birchmeier, W., 1992. A
functional domain in the heavy chain of scatter
factor/hepatocyte growth factor binds the c-Met
receptor and induces cell dissociation but not
mitogenesis, Proc Natl Acad Sci USA, 89: 11574–
11578.
Huff, J. L., Jelinek, M. A., Borgman, C. A., Lansing T. J.,
Parsons JT, 1993. The protooncogene c-sea encodes a
transmembrane proteintyrosine kinase related to the
Met/hepatocyte growth factor/scatter factor receptor,
Proc Natl Acad Sci USA, 90: 6140–6144.
Joung, I. S., Cheatham, T. E., III, 2008. Determination of
alkali and halide monovalent ion parameters for use in
explicitly solvated biomolecular simulations, J. Phys.
Chem. B, 112: 9020–9041.
Levy, Y., Cho, S. S., Onuchic, J. N., Wolynes, P. G., 2005.
A survey of flexible protein binding mechanisms and
their transition states using native topology based
energy landscapes, 346: 1121–1145.
Loncharich, R. J., Brooks, B. R., Pastor, R. W., 1992.
Langevin dynamics of peptides: the frictional
dependence of isomerization rates of N-acetylalanyl-
N'-methylamide, Biopolymers, 32: 523–535.
Montesano, R., Matsumoto, K., Nakamura, T., Orci, L.,
1991. Identification of a fibroblast-derived epithelial
morphogen as hepatocyte growth factor, Cell, 67: 901–
908.
Roe, D. R., Cheatham, T. E., III, 2013. PTRAJ and
CPPTRAJ: software for processing and analysis of
molecular dynamics trajectory data, J. Chem. Theory
Comput., 9: 3084–3095.
Ronsin, C., Muscatelli, F., Mattei, M. G., Breathnach, R.,
1993. A novel putative receptor protein tyrosine kinase
of the met family. Oncogene, 8: 1195–1202.
Ryckaert, J.-P., Ciccotti, G., Berendsen, H. J. C., 1977.
Numerical integration of the Cartesian equations of
motion of a system with constraints: molecular
dynamics of n-alkanes, J. Comput. Phys., 23: 327–341.
Salomon-Ferrer, R., Case, D. A., Walker, R. C., 2013. An
overview of the Amber biomolecular simulation
package, WIREs Comput. Mol. Sci., 3: 198–210.
Salomon-Ferrer, R., Götz, A. W., Poole, D., Le Grand, S.,
Walker, R. C., 2013. Routine microsecond molecular
dynamics simulations with AMBER on GPUs. 2.
Explicit solvent particle mesh Ewald, J. Chem. Theory
Comput., 9: 3878–3888.
Stamos, J., Lazarus R. A., Yao, X., Kirchhofer, D.,
Wiesmann, C., 2004. Crystal structure of the HGF b-
chain in complex with the Sema domain of the Met
receptor, EMBO, 23: 2325–2335.
Tsigelny, I. F., Wheler, J. J., Greenberg, J. P., Kouznetsova,
V. L., Stewart, D. J., Bazhenova, L., Kurzrock, R. 2015.
Molecular determinants of drug-specific sensitivity for
epidermal growth factor receptor (EGFR) exon 19 and
20 mutants in non-small cell lung cancer, Oncotarget,
6: 6029–6039.
SIMULTECH 2016 - 6th International Conference on Simulation and Modeling Methodologies, Technologies and Applications
74
