
Systems Biology Analysis and Literature Data Mining for Unmasking
Pathogenic Neurogenomic Variations in Clinical Molecular Diagnosis
Ivan Y. Iourov
1,2,3
, Svetlana G. Vorsanova
1,2
and Yuri B. Yurov
1,2
1
Mental Health Research Center, Moscow, Russia
2
Separated Structural Unit “Clinical Research Institute of Pediatrics” named after Y. E. Veltishev, Russian National
Research Medical University named after N.I. Pirogov, Ministry of Health of Russian Federation, Moscow, Russia
3
Department of Medical Genetics, Russian Medical Academy of Postgraduate Education, Moscow, Russia
Keywords: Brain Diseases, Clinical Relevance, Genomic Variations, Interpretation Technologies, Molecular Diagnosis,
Neurogenomics, Systems Biology.
Abstract: Biotechnological advances in genomics have significantly impacted on molecular diagnosis. As a result,
uncovering individual genomic variations has made whole-genome analysis attractive for clinical care of
patients suffering from brain diseases. However, to obtain clinically relevant genomic data for successful
molecular genetic/genomic diagnosis, interpretation technologies are recognized to be indispensable. Taking
into account the predictive power of bioinformatics in basic genetic studies, it has been proposed to use in
silico systems biology analysis and data mining for detecting clinically relevant genomic variations by
diagnostic healthcare services. Here, we describe an algorithm used as an integral part of molecular
diagnosis of clinically relevant genomic pathology (neurogenomic variations) in brain diseases. The
bioinformatic technique allows interpreting variations at chromosome and gene levels through systems
biology analysis including literature data mining, which enables to modulate the effect of each genomic
change at transcriptome, proteome and metabolome levels. Studying neurogenomic variations using this
approach, we were able to show that the algorithm can be used as a valuable add-on to whole genome
analysis for diagnostic purposes inasmuch as it appreciably increases the efficiency of molecular diagnosis.
1 INTRODUCTION
Molecular diagnosis of genomic pathology
mediating brain diseases has been appreciably
improved by introducing technologies of whole
genome analysis (i.e. molecular karyotyping and
next-generation sequencing or NGS). The increase
of diagnostic efficiency and new opportunities to
uncover previously unrecognized genetic
mechanisms of brain diseases have led to the wide
use of whole genome scanning techniques (Poot et
al., 2011; Su et al., 2011; Need, Goldstein, 2016;
Anazi et al., 2017). Consequently, this has resulted
into accumulation of huge genomic data sets
requiring new tools for the management (Yurov et
al., 2013, 2017; Iourov et al., 2014). Additionally, in
the neurogenomic context (neurogenomics is defined
as studying the genome for defining
function/malfunction of the nervous system), big
genomic data have been proposed as an empiric
basis of brain research aimed at disease mechanism
discoveries (Boguski, Jones, 2004). Basic studies of
neurogenomic mechanisms of neurodegeneration
and neuropsychiatric diseases have confirmed this
idea and have evidenced such analyses to be almost
inefficient without bioinformatic methods (Iourov et
al., 2009; Yurov et al., 2010, 2013; Heng et al.,
2016). Thus, one can hypothesize that
bioinformatics is also applicable for unmasking
pathogenic neurogenomic variations in molecular
diagnosis.
The application of basic bioinformatic tools in
clinical genomic research has already been proven to
increase the efficiency of molecular genetic
diagnosis (Poot et al., 2011; Xu et al., 2014). For
instance, comparative analyses of original data with
clinical databases (basic data mining) alone is able
to help significantly in interpreting genomic
variaitons (Yen et al., 2017). Studies using more
sophisticated systems biology approaches with
deployed literature data mining show better results
in terms of unmasking clinically relevant genomic
160
Iourov, I., Vorsanova, S. and Yurov, Y.
Systems Biology Analysis and Literature Data Mining for Unmasking Pathogenic Neurogenomic Variations in Clinical Molecular Diagnosis.
DOI: 10.5220/0006649701600165
In Proceedings of the 11th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2018) - Volume 3: BIOINFORMATICS, pages 160-165
ISBN: 978-989-758-280-6
Copyright © 2018 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

variations (Iourov et al., 2014; Dougherty et al.,
2017). Accordingly, the role of bioinformatics in
clinical genome research was highlighted suggesting
in silico interpretation of genomic data to be a
required tool for molecular diagnosis (Heng, Regan,
2017). Our previous studies have formed emprirical
and theoretical basis for developing bioinformatic
interpretational approaches to genome data analysis
in molecular diagnosis of clincially relevant
neurogenomic variations (Vorsanova et al., 2017;
Yurov et al., 2017).
In the present position paper, we propose an
algorithm based on systems biology analysis and
literature data mining for unmasking pathogenic
genomic variations in clinical molecular diagnosis of
brain diseases. We have analyzed original and
previously published data (Yurov et al., 2010, 2013,
2017; Iourov et al., 2014, 2015a, 2015b 2015c;
Vorsanova et al., 2017) to show the extent of
improvement in molecular genetic diagnosis of
clinically relevant neurogenomic variations.
2 ALGORITHM
Bioinformatic analysis based on systems biology
principles is aimed at generation of theoretical
pathways from a genomic variation to a
phenotypical feature. Prior to an in silico systems
biology analysis and literature data mining, there is a
need to possess an appropriate data set to proceed.
The data are usually obtained via multilateral
genome analysis.
It is generally recognized that following datasets
are required to succeed in molecular
genomic/genetic diagnosis: karyotyping data
(chromosomal localization of genomic loci; dataset
required for almost all clinical genetic research);
molecular karyotyping data (copy number
variations; dataset required for uncovering
unbalanced/copy number submicroscopic genome
variations); NGS data (dataset required for detecting
single-gene mutations) (Yurov et al., 2013; Iourov et
al., 2014; Need, Goldstein, 2016; Anazi et al., 2017;
Vorsanova et al., 2017). Figure 1 schematically
overviews a multilateral genome analysis that seems
to be sufficient for an interpretation algorithm based
on in silico systems biology analysis and extended
literature data mining.
Figure 1: Multilateral genome analysis required for
generating data to be evaluated by the algorithm.
Once these data is obtained, genomic variations
are analyzed in the light of previously published
reports and clinical databases (Yen et al., 2017).
However, due to natural limitations of any database
(i.e. impossibility to encompass the complete
variability of the genome in its widest sense),
comparative literature data mining is certainly not
enough for diagnostic purposes (Iourov et al., 2014).
Thus, to succeed in genomic data interpretation,
genes affected by a genomic change should be
functionally assessed by extended literature data
mining and to be analyzed in terms of their
functional significance in epigenomic, proteomic
and metabolomic contexts (Iourov et al., 2014;
Vorsanova et al., 2017; Yurov et al., 2017).
Consequently, it is important to determine
parameters used for such bioinformatics analysis.
Previously, it has been identified that parameters
(i.e. ontology attibuties of genes/proteins or
gene/protein domains) used in in silico evaluations
of genomic rearrangements might be presented as an
absolutely convergent series (Yurov et al., 2017):
where S is finite number of parameters obtained by
data mining and a
i
are integer numbers equal to
numbers of parameters selected for attributing
pathogenic values to a genomic rearrangement.
These parmeters are separated into 4 groups:
genome, epigenome, proteome and metabolome
(Iourov et al., 2014; Yurov et al., 2017). If we
suppose that S does exist for each of these four
In silico systems biology analysis
+
Literature data mining
NGS
Molecular
karyotyping
Karyotyping
Systems Biology Analysis and Literature Data Mining for Unmasking Pathogenic Neurogenomic Variations in Clinical Molecular Diagnosis
161
groups: S
G
is the sum of numbers corresponding to
positive findings of comparative genome data
mining; S
E
is the sum of numbers corresponding to
positive findings epigenome data mining (i.e.
number of brain-specifically expressed genes in
neurogenomic studies); S
I
is the sum of numbers
corresponding to positive associations in interactome
(proteomic analyses of protein-protein interactions)
data mining; S
M
is the sum of numbers
corresponding to positive associations in
metabolome data mining, then pathogenic value of a
genomic variation prioritized through the fusion of
all the aforementioned data sets can be, therefore,
described previously by the inequality (Yurov et al.,
2017):
In other words, if it is possible to identify a
potential effect of a genomic change at one of the
aforementioned levels, the change can be attributed
to an abnormal molecular/cellular process or a
disease pathway (Iourov et al., 2014; Vorsanova et
al., 2017; Yurov et al., 2017). S values equal to zero
or negative S values would correspond to effect lack
and to a positive effect (apparently an extremely rare
condition), respectively. To make possible the
acquisition of these parameters, we have suggested a
pool of procedures for each of four groups and have
orgnaized into an algorithm of interpreting genomic
variation based on systems biology analysis and
literature data mining.
Genomic bioinformatic analysis is performed
through raw data statistical evaluation for excluding
false-positive genome variaitons due to technical
errors and through comparative analysis with
publicly available and/or in-house databases. In
silico epigenomic analysis addresses gene
expression intertissue variability. In silico proteomic
analysis is proposed to be performed by walking
through interactome (interactomic analysis), which
allows to uncover single pathways, pathway clusters
and cryptic ontologies. These pathways can be
further used for candindate proccess identification
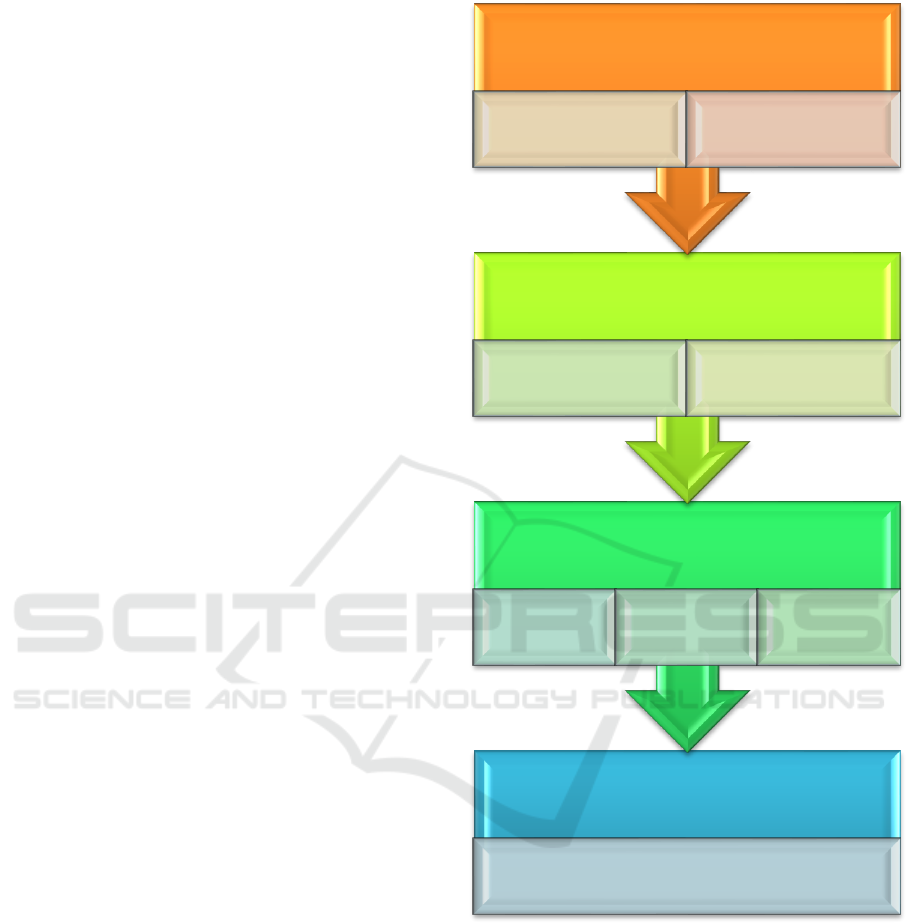
by in silico metabolomic analysis. Figure 2
shematically outlines the algorithm of interpretation
of genome variability.
Figure 2: Schematic overview of the algorithm.
2.1 in Silico Gene Expression — Gene
Prioritization
In silico gene expression analysis has long been
recognized as a tool for gene prioritization (Iourov et
al., 2009, 2014; Satterlee et al., 2015). Nowadays, it
is recognized as a highly useful tool for
neurogenomic (neuroepigenetic) studies (Satterlee et
al., 2015).
In the present algorithm, parameters uncovered
by in silico gene expression analysis are used in
Metabolome
Candidate process identification
Proteome/interactome
Pathway
construction
Ontology-
based process
prioritization
Pathway-
based process
prioritization
Epigenome
Gene prioritization
according to tissue-
specific expression
In silico gene
expression analysis
Genome
Raw data statistical
analysis
Recurrence checking of
neurogenomic
variations
BIOINFORMATICS 2018 - 9th International Conference on Bioinformatics Models, Methods and Algorithms
162

gene prioritization through the distribution of
genomic changes according to tissue-specific
expression variations of the involved genes. Brain-
specific (brain-area-specific) gene expression
represents a set of parameters for subsequent
analysis of genomic variability in the neurogenomic
context (Vorsanova et al., 2017). To be more
precise, positive parameters/values are outlier
expression patterns of genes affected by a genomic
change (i.e. values > 3xM in BioGPS database
http://biogps.org).
2.2 Walking the Interactome
Interactome analysis has recently become a widely
applied technique in the field of genomic and
proteomic bioinformatics. Constructing maps of
protein-protein interactions and their analysis in
terms of ontologies and protein clusterization
according to the involvement in a pathway are able
to give opportunities for pathway-based process
prioritization (Luck et al. 2017). Pathway
involvement and ontologies have been shown as
valuable parameters for in silico evaluations of
functional consequences of neurogenomic
variations, as well (Yurov et al., 2017).
As shown in our previous studies, interactomic
analysis may be a valuable tool for molecular
diagnosis of genome pathology in translational
medicine studies. Owing to the opportunity of
unmasking altered molecular disease pathways by
this bioinformatic approach, the development of
successful molecular-oriented therapeutic
interventions in genetic brain diseases has become
available (Iourov et al., 2015a, 2015c). For instance,
in a previous study, clustering elements of an
interactome built on the basis of molecular
karyotyping data according to pathways has found
useful to delineate altered molecular/metabolic
processes, which were curated by therapeutic
interventions. These interventions have significantly
improved the condition of a patient with subtle
chromosome deletion (Iourov et al., 2015c).
Here, parameters used for the algorithm are
corresponding to numbers of candidate pathways or
processes unraveled in an interactome built
according the results of whole genome analysis.
Generally, a set of genes affected by genomic
changes are proposed to be used for building the
unified interactome. Then, it is possible to determine
clusters of interactome elements according to the
involvement in a pathway or in a molecular process
(i.e. according to ontology).
2.3 in Silico Metabolome Analysis —
Process Prioritization
The algorithm is finalized by in silico metabolome
analysis. This part of the bioinformatics assay
prioritizes processes suggested to be altered by
genomic variations (Yurov et al., 2017). Recently, it
has been shown that bioinformatic systems biology
studies finalized by metabolome/proteome analyses
are key points of clinical, single-cell and
postmortem genomics via pathway-specific profiling
and modeling for defining mechanisms in disease
(Yurov et al., 2010; Wang et al., 2013; Iourov et al.,
2015a, 2015c; Dougherty et al., 2017). Here, these
achievements in basic molecular biology are
proposed to be used in molecular diagnosis of
neurogenomic variations clinically relevant to brain
diseases.
3 THE USE OF THE
ALGORITHM IN CLINICAL
MOLECULAR DIAGNOSIS
Our recent studies have evidenced that in silico
systems biology analysis and extended data mining
for detecting clinically relevant genomic variations
have following benefits: (i) increased yield of
molecular cytogenetic genome analyses (Iourov et
al., 2014); (ii) molecular-oriented therapeutic
interventions in presumably incurable genetic brain
diseases (Iourov et al., 2015c); neurogenomic
disease pathway construction linking genomic
variability and genetic-environmental interactions
(Vorsanova et al., 2017); identification of genomic
causes of pathogenic molecular and cellular
processes (i.e. genome/chromosome instability)
(Iourov et al., 2015a; Yurov et al., 2017). To support
our position paper report on implications of a basic
bioinformatics algorithm used as a valuable add-on
to whole genome analysis for diagnostic purposes,
we have evaluated our data on genomic studies of
children with intellectual disability, autism and
congenital malformations before and after
applications of bioinformatics analysis partially
published before in Iourov et al., 2015b and Iourov
et al., 2016. The results of these evaluations are
depicted by Figure3.
Systems Biology Analysis and Literature Data Mining for Unmasking Pathogenic Neurogenomic Variations in Clinical Molecular Diagnosis
163
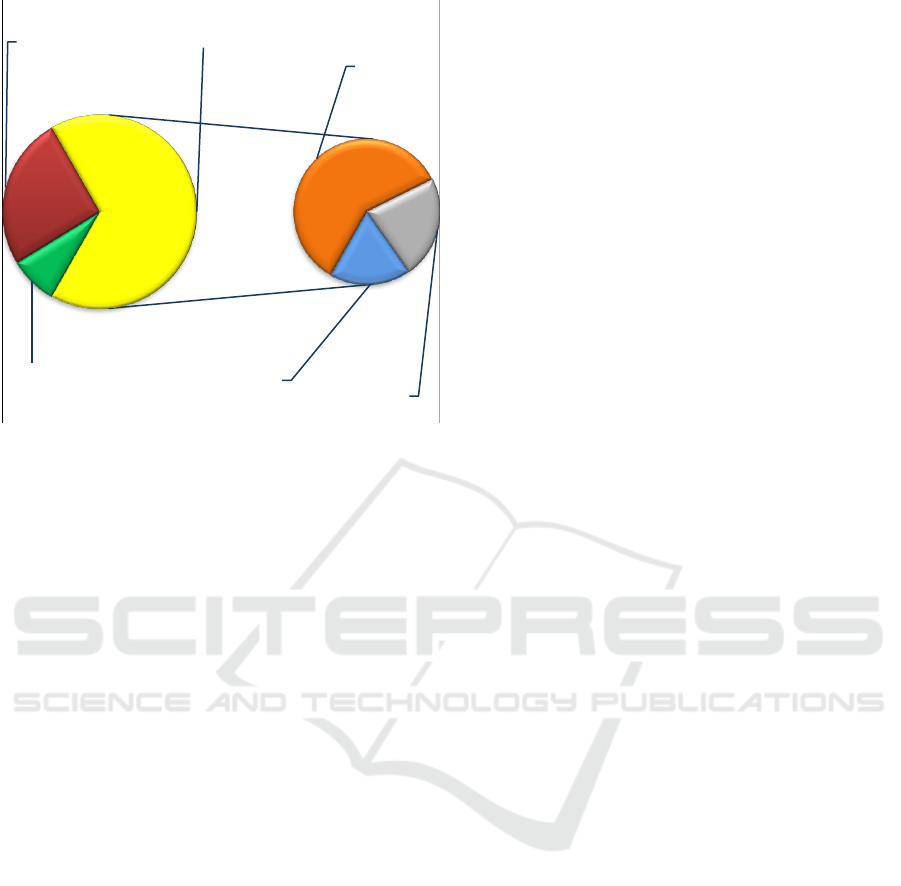
Figure 3: Improvement of molecular neurogenomic
diagnosis by the bioinformatic strategy; red — clinically
relevant neurogenomic variations detected without
bioinformatics; green — neurogenomic variations
clinically irrelevant to the phenotype; yellow — uncertain
results of whole genome analysis without bioinformatics;
orange — clinically relevant multiple neurogenomic
variations confirmed by bioinformatics; grey —
neurogenomic variations resulting in susceptibility to brain
diseases; blue — single gene neurogenomic variations
confirmed by bioinformatics.
As one can see, the application of the algorithm
is virtually able to increase the diagnostic yield. The
efficiency of molecular genome diagnosis with
bioinformatics is 3.6 times higher than that of
genomic analysis lacking bioinformatic
interpretation of neurogenomic variability.
Therefore, one can conclude that bioinformatic
techniques are inseparable from current molecular
diagnosis of neurogenomic pathology with special
attention to disease mechanisms and possible
molecular therapies.
4 CONCLUSIONS
Molecular genetic/genomic diagnosis is consistently
demonstrated to be improved by bioinformatics
approaches. Furthermore, understanding the
functional consequences of genetic variability and
disease mechanisms accomplished by in silico
systems biology evaluations shapes the genome
research making high-resolution genome scans
clinically applicable for any type of disease, at all
ontogenetic stages, in almost all biological
specimens including single cells (Su et al., 2011;
Wan et al., 2013; Yurov et al., 2010, 2013; Satterlee
et al., 2015). In this context, molecular genomic
diagnosis with clinical bioinformatics allows not
only to describe molecular pathology, but also to
become a basis for therapeutic interventions (Iourov
et al., 2015c). In other words, the idea suggesting
that the main issues of personalized medical
genomics might be applicable to specific clinical
tasks (Martin-Sanchez et al., 2004) seems to be
empirically supported.
Finally, the improvement of molecular genomic
diagnosis made through the original bioinformatic
algorithm evidences for the possibility to make
clinical bioinformatics a widely used practice of
healthcare providers. To this end, we suggest that
diagnostic neurogenomics together with clinical
bioinformatics will bring new insights into brain
disease mechanisms and will provide for new
molecular-oriented therapies of currently incurable
conditions.
ACKNOWLEDGEMENTS
This work is supported by ERA.Net RUS Plus
Programme and Russian Foundation for Basic
Research (project: 17-04-01366a). Professors S.G.
Vorsanova and Y.B. Yurov were supported by
Russian Science Foundation (project: 14-15-00411)
during 2014-2016. Professor I.Y. Iourov was
supported by Russian Science Foundation (project:
14-35-00060) during 2014-2016.
REFERENCES
Anazi, S., Maddirevula, S., Faqeih, E., Alsedairy, H.,
Alzahrani, F., Shamseldin, H.E., et al. 2017. Clinical
genomics expands the morbid genome of intellectual
disability and offers a high diagnostic yield. Molecular
Psychiatry 22, 615-624.
Boguski, M.S., Jones, A.R. 2004. Neurogenomics: at the
intersection of neurobiology and genome sciences.
Nature Neuroscience 7, 429-433.
Dougherty, J.D., Yang, C., Lake, A.M. 2017. Systems
biology in the central nervous system: a brief
perspective on essential recent advancements. Current
Opinion in Systems Biology 3, 67-76.
Heng, H.H., Regan, S., Christine, J.Y. 2016. Genotype,
environment, and evolutionary mechanism of diseases.
Environmental Disease 1, 14.
Heng, H.H, Regan, S. 2017. A systems biology
perspective on molecular cytogenetics. Current
Bioinformatics 12, 4-10.
8%
25,5%
39,5%
15%
12%
66,5%
BIOINFORMATICS 2018 - 9th International Conference on Bioinformatics Models, Methods and Algorithms
164

Iourov, I.Y., Vorsanova, S.G., Liehr, T., Kolotii, A.D.,
Yurov, Y.B. 2009. Increased chromosome instability
dramatically disrupts neural genome integrity and
mediates cerebellar degeneration in the ataxia-
telangiectasia brain. Human Molecular Genetics 18,
2656-2669.
Iourov, I.Y., Vorsanova, S.G., Yurov, Y.B. 2014. In silico
molecular cytogenetics: a bioinformatic approach to
prioritization of candidate genes and copy number
variations for basic and clinical genome research.
Molecular Cytogenetics 7, 98.
Iourov, I.Y., Vorsanova, S.G., Demidova, I.A.,
Aliamovskaia, G.A., Keshishian, E.S., Yurov, Y.B.
2015a. 5p13.3p13.2 duplication associated with
developmental delay, congenital malformations and
chromosome instability manifested as low-level
aneuploidy. SpringerPlus 4, 616.
Iourov, I.Y., Vorsanova, S.G., Korostelev, S.A., Zelenova,
M.A. and Yurov, Y.B., 2015b. Long contiguous
stretches of homozygosity spanning shortly the
imprinted loci are associated with intellectual
disability, autism and/or epilepsy. Molecular
cytogenetics, 8, 77.
Iourov, I.Y., Vorsanova, S.G., Voinova, V.Y., Yurov,
Y.B. 2015c. 3p22.1p21.31 microdeletion identifies
CCK as Asperger syndrome candidate gene and shows
the way for therapeutic strategies in chromosome
imbalances. Molecular Cytogenetics 8, 82.
Iourov IY, Vorsanova SG, Korostelev SA, Vasin KS,
Zelenova MA, Kurinnaia OS, Yurov YB. 2016.
Structural variations of the genome in autistic
spectrum disorders with intellectual disability. Zhurnal
Nevrologii i Psikhiatrii imeni S.S. Korsakova. 116(7),
50-54.
Luck K, Sheynkman GM, Zhang I, Vidal M. 2017.
Proteome-scale human interactomics. Trends in
Biochemical Sciences 42, 342-354.
Martin-Sanchez, F., Iakovidis, I., Nørager, S., Maojo, V.,
de Groen, P., Van der Lei, et al. 2004. Synergy
between medical informatics and bioinformatics:
facilitating genomic medicine for future health care.
Journal of Biomedical Informatics 37, 30-42.
Need, A.C., Goldstein, D.B. 2016. Neuropsychiatric
genomics in precision medicine: diagnostics, gene
discovery, and translation. Dialogues in Clinical
Neuroscience 18, 237-252.
Poot, M., Van Der Smagt, J.J., Brilstra, E.H., Bourgeron,
T. 2011. Disentangling the myriad genomics of
complex disorders, specifically focusing on autism,
epilepsy, and schizophrenia. Cytogenetic and Genome
Research 135, 228-240.
Satterlee, J.S., Beckel-Mitchener, A., Little, A.R.,
Procaccini, D., Rutter, J.L., Lossie, A.C. 2015.
Neuroepigenomics: resources, obstacles, and
opportunities. Neuroepigenetics 1, 2-13.
Su, Z., Ning, B., Fang, H., Hong, H., Perkins, R., Tong,
W., Shi, L. 2011. Next-generation sequencing and its
applications in molecular diagnostics. Expert Review
of Molecular Diagnostics 11, 333-343.
Vorsanova, S.G, Yurov, Y.B, Iourov, I.Y. 2017.
Neurogenomic pathway of autism spectrum disorders:
linking germline and somatic mutations to genetic-
environmental interactions. Current Bioinformatics 12,
19-26.
Wang, Q., Zhu, X., Feng, Y., Xue, Z., Fan, G. 2013.
Single-cell genomics: An overview. Frontiers in
Biology 8, 569-576.
Xu, F., Li, L., Schulz, V. P., Gallagher, P. G., Xiang, B.,
Zhao, H., Li, P. 2014. Cytogenomic mapping and
bioinformatic mining reveal interacting brain
expressed genes for intellectual disability. Molecular
Cytogenetics 7, 4.
Yen, J. L., Garcia, S., Montana, A., Harris, J., Chervitz, S.,
Morra, M., West, J., Chen, R., Church, D. M. 2017. A
variant by any name: quantifying annotation
discordance across tools and clinical databases.
Genome Medicine, 9, 7.
Yurov, Y.B., Vorsanova, S.G., Iourov, I.Y. 2010.
Ontogenetic variation of the human genome. Current
genomics 11, 420-425.
Yurov, Y.B., Vorsanova, S.G., Iourov, I.Y. 2013. Human
interphase chromosomes. Springer, New York, NY.
Yurov, Y.B., Vorsanova, S.G., Iourov, I.Y. 2017.
Network-based classification of molecular cytogenetic
data. Current Bioinformatics 12, 27-33.
Systems Biology Analysis and Literature Data Mining for Unmasking Pathogenic Neurogenomic Variations in Clinical Molecular Diagnosis
165
