
Bio-backfill: A Scheduling Policy Enhancing the Performance of
Bioinformatics Workflows in Shared Clusters
Ferran Badosa
1
, Antonio Espinosa
1
, Gonzalo Vera
2
and Ana Ripoll
1
1
Computer Architecture and Operative Systems Department of Universitat Aut
`
onoma de Barcelona, Bellaterra, Spain
2
Center for Research in Agricultural Genomics, Bellaterra, Spain
Keywords:
Bioinformatics Workflows, Slowdown, Dependencies, Scheduling Policies, Backfill, Resource Management
Systems.
Abstract:
In this work we present the bio-backfill scheduler, a backfill scheduler for bioinformatics workflows appli-
cations running on shared, heterogeneous clusters. Backfill techniques advance low-priority jobs in cluster
queues, if doing so doesn’t delay higher-priority jobs. They improve the resource utilization and turnaround
achieved with classical policies such as First Come First Served, Longest Job First.. When attempting to
implement backfill techniques such as Firstfit or Bestfit on bioinformatics workflows, we have found several
issues. Backfill requires runtime predictions, which is particularly difficult for bioinformatics applications.
Their performance varies substantially depending on input datasets and the values of its many configuration
parameters. Furthermore, backfill approaches are mainly intended to schedule independent, rather than depen-
dent tasks as those forming workflows. Backfilled jobs are chosen upon its number of processors and length
runtime, but not by considering the amount of slowdown when the Degree of Multiprogramming of the nodes
is greater than 1. To tackle these issues, we developed the bio-backfill scheduler. Based on a predictor generat-
ing performance predictions of each job with multiple resources, and a resource-sharing model that minimizes
slowdown, we designed a scheduling algorithm capable of backfilling bioinformatics workflows applications.
Our experiments show that our proposal can improve average workflow turnaround by roughly 9% by and
resource utilization by almost 4%, compared to popular backfill strategies such as Firstfit or BestFit.
1 INTRODUCTION
Advancements in computing and biology have re-
duced the cost and time of genome sequencing by
80% over the past ten years (NHGRI, 2017). As a
consequence, the size of genomic databases, many
of which are available worldwide, has grown increas-
ingly. To analyze the ever-increasing volumes of ge-
nomic data, bioinformatics applications can be em-
ployed. Analysis allows for extraction of valuable in-
formation regarding subjects’ propensity to develop
certain deceases or response to drugs. Thanks to that
knowledge, individualized medical treatments can be
developed. Many kinds of bioinformatics applica-
tions exist, performing different tasks involved in
genome analysis, such as genome sequencing, align-
ment or annotation. Users of such applications, usu-
ally data analysts or biologists, may build bioinfor-
matics workflows by combining multiple applications
in a defined structure. Aside from input and output
data, workflows also have intermediate data. That is,
output data generated by a precedent application (A)
which is in turn input data of another, posterior appli-
cation (B). Application B must wait until A terminates
in order to get the necessary data to start execution.
Unlike independent tasks, which are always in ready
status for execution, workflows applications may have
dependencies. Non-ready applications such as B may
be further defined by specifying the remaining time
for its dependencies to be solved.
Bioinformatics applications are usually highly
resource-demanding. Clusters formed by hetero-
geneous nodes with multiple cores and multiple
sockets have become common platforms to execute
them, since they provide cost-effective access to vast
amounts of resources. Before submitting applications
to heterogeneous clusters, users must determine the
resources needed by their applications. This is a hard
task even when dealing with applications whose per-
formance barely changes from one execution to the
next.
When dealing with bioinformatics applications, a
148
Badosa, F., Espinosa, A., Vera, G. and Ripoll, A.
Bio-backfill: A Scheduling Policy Enhancing the Performance of Bioinformatics Workflows in Shared Clusters.
DOI: 10.5220/0006812901480156
In Proceedings of the 3rd International Conference on Complexity, Future Information Systems and Risk (COMPLEXIS 2018), pages 148-156
ISBN: 978-989-758-297-4
Copyright
c
2019 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

higher degree of difficulty is faced. The resource
requirements and execution times of bioinformatics
workflows applications may vary substantially from
one execution to the next. Performance variation of
applications depends on two factors. First, the char-
acteristics of input data selected for analysis, such as:
the size of the input dataset file or files, the number
of sequences within, or its length. Second, the val-
ues of the multiple configuration parameters each ap-
plication has, which must be declared by users. By
giving different values to these parameters, users may
conduct different kinds of analysis on the same input
datasets. Some analysis seek high-quality results, re-
quiring resource-demanding and long-lasting compu-
tations. Conversely, other analysis only target lower-
quality results, which are much easier and quicker to
obtain. The large amount of existing combinations
of data and parameters values, and the performance
variations they may cause on applications, makes it
additionally difficult to determine the resources or ex-
ecution times of a single application.
For the present work, we considered bioinfor-
matics applications programmed in shared-memory
paradigms. Their execution is conducted by multi-
ple execution threads running in a single node at a
time. Shared-memory paradigms offer numerous ad-
vantages for bioinformatics applications. They sim-
plify users’ job submissions, requiring little or no pre-
vious knowledge of computing environments. More-
over, they allow threads to easily communicate by us-
ing the memory. Hence, through high-speed paths,
and requiring no external synchronization mecha-
nisms. Many bioinformatics applications are pro-
grammed in shared-memory paradigms.
One major category of bioinformatics applica-
tions is that of read mappers, which can be employed
to analyze reads sequences, that is, fragments of a
partially-sequenced genome. First though, the posi-
tion in the genome of the base pairs of each read se-
quence must be found. To do so, mappers compare
each sequence in the reads file against each sequence
of a fully-sequenced reference genome, which is used
as a template. These input files may amount up to
tens or hundreds of gigabytes and contain millions of
sequences each. Mappers, as many bioinformatics ap-
plications, are usually labeled as data-intensive. Map-
ping algorithms usually rely on large memory capaci-
ties in order to analyze such sizable genomic datasets.
Execution threads request a great number of mem-
ory accesses as mapping algorithms compare each
pair of reference-reads sequences. Handling these
amounts of requests has been a major concern of nu-
merous research articles (Waidyasooriya et al., 2014;
Xin et al., 2013), yet it still poses a major chal-
lenge. If the number of threads is increased, more
sequences are simultaneously compared, intensifying
the amount of memory requests. The large num-
ber of requested memory accesses may overwhelm
nodes’ bandwidth, leading to saturation and length-
ening latencies. As a result, Processing Units (PUs)
may remain idle or low utilized as they wait for re-
quested data. Due to that, some read mapping ap-
plications show limited scalability with a low num-
ber of PUs (Al-Ali et al., 2016; Kathiresan et al.,
2014). This phenomenon must be considered when
scheduling mappers to avoid compromising their per-
formance and that of the system. Popular mapping
or alignment applications are blast or BWA (Li and
Durbin, 2009), which employ mapping algorithms
such as Needleman-Wunsch (Needleman and Wun-
sch, 1970) or Burrows Wheeler Transform (Burrows
and Wheeler, 1994).
Mappers are mainly regarded as memory-bound
applications. However, depending on the data char-
acteristics and parameters values, that situation may
be reversed, causing mappers to become bounded by
other resources such as cpu.
Another category of bioinformatics applications is
that formed by phylogenies. They study the evolution
of genetically-related organisms by building a phy-
logeny three. Common methods to compute trees in-
clude maximum likelihood estimations or Bayesian
inferences. Examples of phylogenies are mrbayes
(Huelsenbeck and Ronquist, 2001) or phyml (Guin-
don and Gascuel, 2003). Unlike mappers, phyloge-
nies are generally bounded by the cpu.
Most clusters are shared among multiple work-
flow applications, which compete for resources. The
availability of resources varies over time upon the ex-
ecution times and resource usage of running appli-
cations. Depending on resource availability, needs
and priorities, some jobs are granted access to certain
amounts of resources, whereas others have to wait.
In shared clusters, multiple jobs may share pro-
cessors of the same node. Thus, the Degree of Multi-
programming of the nodes (DP) may be greater than
one. Sharing resources may be beneficial for overall
performance, decreasing waiting times and increas-
ing resource utilization. Nonetheless, competition for
same-node resources may cause jobs to slow down
their execution times (Figueira and Berman, 2001).
The slowdown extent depends on the resource usage
made by jobs sharing the same nodes. However, it
can be minimized if combinations of jobs are prop-
erly scheduled in the different nodes. That is, com-
bining for instance memory-bound applications with
cpu-bound applications.
Resource Management Systems (RMS) admin-
Bio-backfill: A Scheduling Policy Enhancing the Performance of Bioinformatics Workflows in Shared Clusters
149

ister resources. They monitor the queue of jobs
and system status, in order to schedule those re-
sources. RMS schedulers determine the priorities and
resources of jobs based on their own scheduling al-
gorithms. Scheduling algorithms can be divided into
time-sharing and space-sharing. Time sharing algo-
rithms divide processing time into to slots, and assign
them to pending jobs. Space-sharing algorithms are
the most common ones implemented in schedulers.
They allocate resources to jobs until execution fin-
ishes, reducing overhead. Examples of space-sharing
algorithms are: FCFS (First Come First Serve), SJF
(Shortest Job First), LJF (Longest Job First) or EDF
(Earliest Deadline First).
When these policies are implemented on a queue
of jobs, scheduling gaps may be generated. That
may cause processors to remain idle over time, as
other jobs wait. To fill the scheduling gaps, the
aforementioned policies can be combined with back-
fill. Backfill increases the initial priorities of queued
jobs, and fits them on idle processors, as long as do-
ing so doesn’t delay the expected start time of any
higher-priority jobs. Applying backfill enhances re-
source utilization and turnaround. Resources that
would otherwise remain idle are kept busy by back-
filled jobs. As previously mentioned, determining
the resources needed by a single bioinformatics ap-
plication is difficult since they may vary substantially
depending on user-selected combination of data and
parameters values. When multiple workflows share
the same cluster, with variable resource availability
and job queues, such uncertainty may lead to naive
scheduling approaches being taken, i.e. over allo-
cation of resources. In those cases, longer waiting
times arise, whereas low utilization of resources is at-
tained. Resources may go wasted and users can see
their turnaround times extended.
Backfill techniques may be suitable to properly
schedule bioinformatics workflows applications and
improve overall system performance. Nonetheless,
we have found three major drawbacks when review-
ing current backfill techniques. First, backfill requires
execution time predictions to be provided, since the
start time of queued jobs depends on completion time
of previous jobs. Providing execution time predic-
tions of bioinformatics applications may be hard or
unfeasible, due to their variable performance, which
depends on parameters and data. The second draw-
back we found is that when current backfill tech-
niques advance jobs, they don’t consider the execu-
tion time slowdown caused by the different combina-
tions of applications, which can also have significant
repercussions on the eventual turnaround times.
The third drawback is that backfill techniques
mainly designed for being applied on independent
tasks, and rarely feature within workflow scheduling
environments (Wu et al., 2015). Although recent ef-
forts have been made (Arabnejad et al., 2017) on the
matter, these fail to adapt to the particular variable-
resource needs of bioinformatics workflows applica-
tions.
To face these drawbacks, we present the bio-
backfill scheduler, a novel technique for scheduling
bioinformatics workflows applications in shared clus-
ters, considering their parameter values, data charac-
teristics, and dependencies. The bio-backfill sched-
uler is based on a pre-scheduling framework, re-
viewed in Section 3.1. The pre-scheduling frame-
work includes a historical database and a prediction
model, which automatically generates jobs’ perfor-
mance predictions with different resources. Thanks
to predictions, dependency times can also be calcu-
lated. The bio-backfill scheduler, reviewed in Section
3.2, includes a resource-sharing model that consid-
ers the slowdown spawned when the DP of the nodes
is greater than one. That is, determines which jobs
are most compatible for same-node execution so that
slowdown is minimized. In Section 4, we process
a series of bioinformatics workflows on a heteroge-
neous cluster with the bio-backfill scheduler, as well
as with other backfill policies. Experiments prove that
the proposed bio-backfill scheduler can improve the
average turnaround and resource utilization of bioin-
formatics workflows applications obtained with other
backfill approaches.
2 RELATED WORK
Among the main current backfill techniques applied
in clusters we can find Firstfit backfill, Bestfit back-
fill, Greedy backfill or Preemptive backfill. The first
step in most common backfill techniques is similar.
The queue of jobs, which has already been scheduled
resources and set priorities with other policies (i.e.
FCFS, Shortest Job First, Longest Job First...), is fil-
tered. The list of potential backfill candidates, formed
by those jobs fitting in the current backfill window, is
extracted. The second step is different depending on
the backfill technique applied. Firstfit backfill consid-
ers all backfill candidates, and selects and starts the
first one. Bestfit calculates the degree of fit of each
job, based on different backfill metrics: the number
of processors, the execution time in seconds, or the
product of both. Next, starts the job with the best
fit. Greedy backfill assesses the degree of fit of each
combination of candidate jobs for backfilling, based
on the same metrics than Bestfit. All jobs in the best
COMPLEXIS 2018 - 3rd International Conference on Complexity, Future Information Systems and Risk
150

combinations are started. Finally, Preemptive backfill
jobs are given priorities, based on different parame-
ters: current running duration, number of processors
on which job runs.. Preemptive backfill then starts
the highest-priority candidate. All techniques iterate
over steps one and two, as long as there are remaining
backfill candidates and idle resources.
Backfill techniques advance jobs if the expected
start time of any higher-priority jobs is not delayed.
However, that constraint can be relaxed, and differ-
ent advancing criteria can be chosen, such as done by
conservative backfilling, aggressive or easy backfill-
ing, and slack backfilling. In conservative backfill-
ing, any job is given resource reservation as it arrives
in the queue. Backfill candidates can be then back-
filled if no higher-priority jobs see their expected start
time delayed. In easy backfilling, only the first job
of the queue is given resource reservation. Backfill
candidates are then granted permission to advance as
long as they don’t delay the start of the first, resource-
reserved job.
No clear criteria is defined on which technique,
conservative or aggressive backfill, is better than the
other. Conservative backfilling allows less jobs to be
backfilled than aggressive backfilling. Since there are
more jobs whose starting time must be considered,
more constraints exist in order for other jobs to be
backfilled. Furthermore, in conservative backfilling
there may be multiple jobs with reserved resources,
and thus may become hard to fit more backfilled jobs.
Conversely, in easy backfilling which there’s only one
job with reserved resources, and more jobs can be eas-
ily backfilled.
Easy or conservative backfilling may favor ear-
lier execution of jobs, depending on their character-
istics, such as length (duration) and width (number
of processors). Conservative backfilling reserves re-
sources for more jobs, spawning earlier execution of
short wide jobs. These jobs have smaller chances of
being backfilled since the more processors a jobs, the
more difficult it is to fit them into scheduling gaps.
With easy backfilling, short wide jobs might have to
wait until they get on top of the queue to get a reser-
vation of resources, increasing waiting times. Con-
versely, long narrow jobs may start earlier with easy
backfilling than with conservative backfilling. Due to
little processors being required, they can be relatively
easy be fit into gaps. As for short narrow jobs and
long wide jobs, it is harder whether they may be ben-
efited by easy or conservative backfilling. Generally,
they may equally as favored by initial reservation as
by backfilled reservation.
Slack backfill is based on conservative backfilling,
but it is more flexible since allows certain delays of
jobs, called slacks (Talby and Feitelson, 1999). De-
pending on its priority, each job may be given a dif-
ferent slack, determining how long it may have to wait
before starting execution. Any job may be backfilled
if doing so doesn’t delay any other jobs by longer
than their slack. Results show that slack backfill re-
duces average waiting time by 15% compared with
easy backfill, under identical conditions (Bucur and
Epema, 2001).
Multiple-queue backfill (Lawson and Smirni,
2002) is based on aggressive backfill. Input jobs are
monitored, and according to the length of their exe-
cution time estimations, rearranged in different wait-
ing queues. The system is divided into variable par-
titions with equal amount of processors, and each
queue is assigned a partition. Nevertheless, if a job
of one partition remains idle, it can be used by jobs
in another partition. Consequently, the partition sizes
of the system are dynamic, and processors are ex-
changed among partitions based upon load. For multi-
ple queues, narrow jobs are executed earlier than wide
jobs. The main strength of multiple-queue policies is
that reduces the likelihood of short jobs getting de-
layed in the queue behind the long ones.
Although many backfill mechanisms exist, ones
favoring jobs by characteristics: length, width or
combination of both, after reviewing the literature,
we haven’t found any adapted to the particularities of
bioinformatics applications. Namely, those particular
characteristics upon which its width and length
depend on such great extent: input dataset and pa-
rameters values. Also, it is important to point out that
current backfill techniques don’t consider the amount
execution time slowdown spawned when multiple
applications share the same node. Hence, we propose
a new bio-backfill approach, described in Section 3,
which accounts for both factors. Furthermore, unlike
many backfill algorithms, the proposal is intended
for workflows. It includes accountability of tasks’
status, ready or non-ready, as well as the predicted
remaining time for tasks to become ready.
3 PROPOSAL: BIO-BACKFILL
SCHEDULER
In this work we present the bio-backfill scheduler,
a new backfilling approach for bioinformatics work-
flows applications running in shared heterogeneous
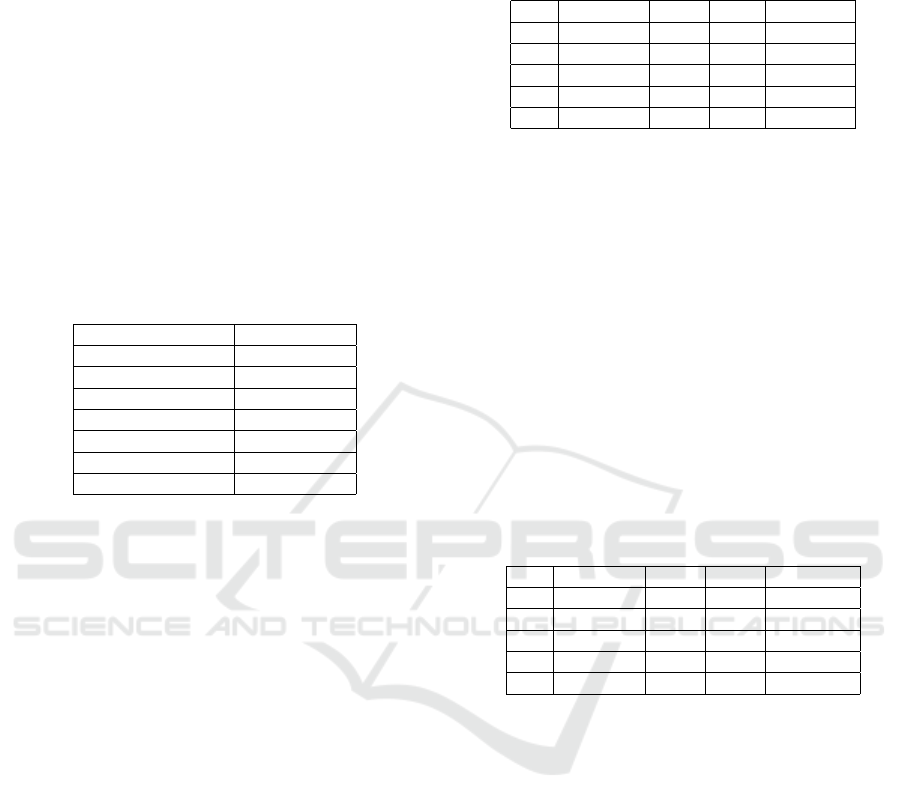
clusters. The proposal, depicted in Figure 1, is formed
by a pre-scheduling framework (database and predic-
tor), and a bio-backfill scheduling algorithm. The
framework is reviewed in Section 3.1, whereas the
Bio-backfill: A Scheduling Policy Enhancing the Performance of Bioinformatics Workflows in Shared Clusters
151

bio-backfill scheduler, main scope of this work, is ex-
plained in Section 3.2.
Figure 1: Proposed bio-backfill scheduler, including the
pre-scheduling framework, formed by a database and a pre-
dictor.
3.1 Pre-scheduling Framework:
Historical Database and Prediction
Model
The bio-backfill scheduler is based on a pre-
scheduling framework, mainly formed by a histori-
cal database and a multivariate regression predictor.
Since the main focus of this work is the bio-scheduler,
the pre-scheduling framework will be briefly re-
viewed. The framework is based on earlier research
work (Badosa et al., 2017), which may be consulted
for further information.
To develop a predictor, the performance of jobs
executed in the cluster must be previously stored
and analyzed. Hence, we developed a methodol-
ogy to track the performance of each executed job.
Namely, we monitored resource metrics such as mem-
ory and CPU consumptions and usage patterns, as
well as turnaround. A variety of Linux tools were
used: /usr/bin/time, sar, pidstat, perf, vmstat.. At
jobs’ completion, performance information is stored
in the historical database. Equation 1 shows a sim-
plified representation of stored information for each
Job
ID
= app, params, data.
Per f Job
ID
= app, params, data, resources,time (1)
As previously mentioned, backfilling approaches
require users to provide accurate runtime predictions
at submission time. That may become challenging for
users of bioinformatics applications, due to its vari-
able performance, highly influenced by parameters
and data. One of the advantages of the bio-backfill
proposal is that relieves users of having to provide
time estimations. Instead, the Multivariate Regression
Predictor automatizes this process.
To build the Multivariate Regression Predictor,
database information was statistically analyzed. The
relevant predictor variables were determined with
the Pearson Correlation Coefficients. To prevent
the model from becoming over-fitted, quality metrics
such as Adjusted R
2
, were used. The accuracy of the
prediction, was assessed with the Mean Relative Error
metric. Once built, the predictor generates for each
submitted Job
ID
, multiple performance predictions
with different combinations of resources, i.e., in each
node, with a range of PUs. With the multiple predic-
tions received per job, the bio-backfill scheduler can
determine which set of resources suit each jobs’ needs
under different scenarios of resource availability. Fur-
thermore, predictions with different resources allow
for the scheduler to calculate speedups and efficien-
cies of each job. The threshold of PUs beyond which
jobs’ execution time doesn’t decrease (PU
MaxSpeed
),
or the execution time penalties when running with less
PUs: (T
Pen
=(PU
MaxSpeed
)-(PU
LowSpeed
), can be calcu-
lated. This way, the trade off between speed and effi-
ciency of shared clusters can be properly dealt with.
Bioinformatics applications can be executed under
many combinations of parameters and data in the het-
erogeneous cluster. To generate reliable and accurate
predictions, large amounts of data, stemming from
numerous executions, should be gathered. The bio-
backfill proposal includes a feedback mechanism that
harnesses performance information of every job exe-
cuted by users in the cluster. Once a job finishes, new
performance information is added to the database.
Next, the difference between real and predicted re-
sults is calculated. This way, the prediction model can
be continuously updated with newly-obtained perfor-
mance results. The feedback is depicted in Figure 1,
with a dashed line.
3.2 Bio-backfill Scheduler
As previously discussed, low scalability shown by
some read mappers may cause a part of node re-
sources such as PUs to go wasted. Backfill algorithms
can allocate these PUs to other waiting jobs, increas-
ing resource utilization and reducing turnaround. De-
pending on their algorithms, schedulers filter a series
of backfill candidates, that is those jobs whom if ad-
vanced, won’t delay start time of high priority jobs. In
this section we review the scheduling algorithm that
we developed and included in the bio-backfill sched-
uler. A summarized pseudo-code version of the algo-
rithm is shown in Algorithm 1.
To select which of the backfill candidates is to
be chosen, several parameters can be accounted for,
such as candidates’ width (number of processors) or
length (predicted runtime). However, many back-
fill techniques don’t consider the slowdowns on jobs’
makespans caused by the different potential combina-
tions of jobs to share the same nodes. That is, when
COMPLEXIS 2018 - 3rd International Conference on Complexity, Future Information Systems and Risk
152

the DP>1. In this work we calculate makespan slow-
down of Job sharing a node with a Load, as in Equa-
tion 2, in percentages.
Slowdown
Job,Load
=
|Mak.
Job,Excl
− Mak.
Job,Shared
|
Makespan
Job,Excl.
(2)
Jobs requesting similar node resources throughout
their executions will likely increase slowdown much
longer than combinations of jobs requesting different
resources. In turn, this will cause other, non-requested
resources to go wasted, reducing utilization and in-
creasing waiting times.
The scheduling algorithm backfills candidates not
only upon its width or length, but also considering
the different slowdowns caused by each candidate
when simultaneously running with the nodes’ cur-
rent loads. Hence, the bio-backfill scheduling algo-
rithm is capable of minimizing the slowdown when
the DP>1. This feature further enhances performance
improvements achieved by current strategies, which
may schedule non-compatible jobs in the same nodes.
As mentioned in Section 3.1, the feedback mechanism
of the bio-backfill scheduler stores performance infor-
mation of each execution. Out of this information, the
amount of slowdown in shared mode can be obtained.
Unlike many backfill schedulers, intended for
scheduling independent tasks, the bio-backfill sched-
uler has been designed for scheduling bioinformat-
ics workflows applications. That is, tasks that aren’t
always ready but may have dependencies. Hence,
we considered the status of each workflow task:
Ready/NonReady, along with the predicted time for a
tasks’ dependencies to be solved WaitDep. WaitDep
will depend on the precedent tasks, their execution
times or own dependencies, and ongoing scheduling
policy. The execution times of precedent tasks can
be predicted thanks to the predictor included in the
pre-scheduling framework. From these predictions,
the waiting time for each tasks’ dependencies to be
solved, WaitDep, can be predicted too.
The algorithm is fed with four inputs, as can be
seen in the top of the pseudo-code. First, the List of
queued Jobs to schedule (LJobs), each with its pa-
rameters and data. Second, the List of Performance
Predictions of jobs, calculated in each node of the
cluster with a range of PUs. Third, the List of De-
pendency Status (LDep) of every job in LJobs, de-
termining whether each job in LJobs is either ready
or NonReady, and WaitDep times. Fourth, the List
of Slowdowns (LSlow) which states the slowdown
times caused when scheduling different combinations
of jobs in LJobs for same-node execution. With these
inputs, the algorithm is capable of determining the re-
sources and priorities of jobs so that average workflow
turnaround is minimized, and resource usage maxi-
mized. The algorithm generates a priority-sorted List
of scheduled jobs (LPrio).
In shared environments with variable avail-
ability, waiting jobs may not be able to run
with as much resources (MaxRes) as to maximize
speedup (PU
MaxSpeed
), but have to run with less
resources (AvailRes), lengthening execution times
(PU
LowSpeeds
). However, it may occur that in a near
future, a job finishes, releasing enough resources as
for the waiting job to run with PU
MaxSpeed
. In these
cases, the scheduler can opt between executing the job
at once with AvailRes, or waiting for MaxRes release
and execute the job with PU
MaxSpeed
. To minimize
turnaround, our scheduler compares both options and
proceeds with the fastest one. Sometimes, a queued
job which is not ready (but will be shortly), may be
much more compatible with a node’s current load
than ready jobs. In these cases it may be favorable for
overall performance to wait for that job to go ready,
and schedule it alongside the current load, instead of
scheduling at once the most compatible ready task.
In these cases, the algorithm compares the WaitDep
time of the highly-compatible shortly-ready task plus
the its slowdown, with the slowdown of the currently-
ready task. By comparing these two amounts of times,
the bio-backfill algorithm determines whether wait-
ing WaitDep compensates or not. Similarly, when
scheduling combinations of jobs in the same node, the
algorithm schedules the job with longest average pre-
dicted time with PU
MaxSpeed
, alongside the most com-
patible ready job. Also, it calculates the slowdowns of
the longest job alongside non-ready jobs. Then com-
pares the minimum slowdown of ready jobs (Slow
Re
),
with the minimum sum of: WaitDep times plus slow-
downs of non ready jobs (Wait&Slow
NoRe
), in order
to decide whether it must wait.
4 EXPERIMENTS
In this section we test the bio-backfill scheduling pro-
posal, and compare it with other relevant backfill ap-
proaches in the literature. To do so, we simulate the
processing of a series of synthetic workflows on a
cluster partition, described below. Workflows are pro-
cessed 3 times, each with different backfill policy: the
proposed Bio-backfill scheduler, Firstfit, and Bestfit,
respectively. At the end, turnarounds and resource
utilizations obtained with the three policies are com-
pared and discussed. The layout of the experiments is
depicted in Figure 2.
Bio-backfill: A Scheduling Policy Enhancing the Performance of Bioinformatics Workflows in Shared Clusters
153
Algorithm 1: Scheduling pseudocode summary.
Inputs : LJobs: List of queued Jobs (Job
ID=1,..,q
=app,param,data)
LPred: List of Perf.Preds. for PUs in node, for job in LJobs
LDep: List of Dependency Status for job in LJobs
LSlow: List of Slowdowns, for job in LJobs
Output: LPrio: Priority-Sorted List of Jobs
1 while jobs in LJobs do
; // For each Job
ID
, in different res.(node,PU)
2 Calculate Perf.Preds. (LPred); // times,speedups,effi.
3 Calculate Slowdowns (LSlow); // dif.combis of jobs
4 Calculate Job Dependencies (LDep); // Pred. WaitDep
5 MaxRes
ID
= PUs for Max SpeedUp ; // for Job
ID
6 LowRes
ID
= Range PUs below MaxRes
ID
; // t.penalties
7 Read Resource Status; // res avail, nodes’ load
8 if IdleNodes in cluster then
9 Job
LRe
= Longest Ready Job
ID
10 Slow
Re
= min Slow (Job
OthersRe
,Job
LRe
)
11 Wait&Slow
NoRe
=Slow(Job
OthersNoRe
,Job
LRe
)+WaitDep
12 SelectedJobs= Jobs with min (Slow
Re
,WaitSlow
NoRe
) if
ResAvail(node) < MaxRes(SelectedJobs) then
13 SelecRes = PUs minimizing sum of LPred times
14 end
15 else
16 SelectRes = MaxRes for SelectedJobs
17 end
18 end
19 if LoadedNodes in cluster then
20 Slow
Re
= MinSlow(Job
Load
,Job
ID.Re
)
21 Wait&Slow
NoRe
=min (Slow(Job
Load
,Job
ID.Re
)+WaitDep)
22 SelectedJobs = Jobs with min(Slow
Re
; Wait&Slow
NoRe
)
23 if ResAvail(node) > MaxRes(SelectedJobs) then
24 SelecRes = MaxRes for SelectedJobs
25 end
26 else
27 Time
AvailRes
= PredTime Job
ID,AvailRes
28 Wait&Time
MaxRes
= Wait
MaxResFree
+ Time
MaxRes
29 SelectedRes=min(Time
AvailRes
,Wait&Time
MaxRes
)
30 end
31 end
32 end
33 return List of Jobs sorted by Priority (LPrio)
Figure 2: Assessing the performance of different backfill
policies, proposed Bio-backfill, Firstfit and Bestfit, on a set
of workflows.
4.1 Workload Definition and
Performance Data Generation
The first step consisted in defining a set of syn-
thetic workflows whose processing will be simu-
lated with the mentioned backfill policies. Work-
flows applications have been chosen upon relevance
reported by benchmarks (Hatem et al., 2013; Lord
et al., 2015). Namely, we selected cpu-bound align-
ers and mappers: blast 2.6.0 (Altschul et al., 1990),
bwa-mem and bwa-aling 0.7.5a (Li and Durbin,
2009), bowtie 2.2.6 (Langmead, 2009), soap 2.21 (Li
et al., 2008), star 2.4.2a (Dobin et al., 2013), hisat
2.0.5 (Kim et al., 2015), and cpu-bound phyloge-
nies: phyml 2.4.5par (Guindon and Gascuel, 2003),
mrbayes 3.1.2h (Huelsenbeck and Ronquist, 2001),
raxml 8.2.9 (Stamatakis et al., 2004) and fasttree
2.1.3.c (Price et al., 2009).
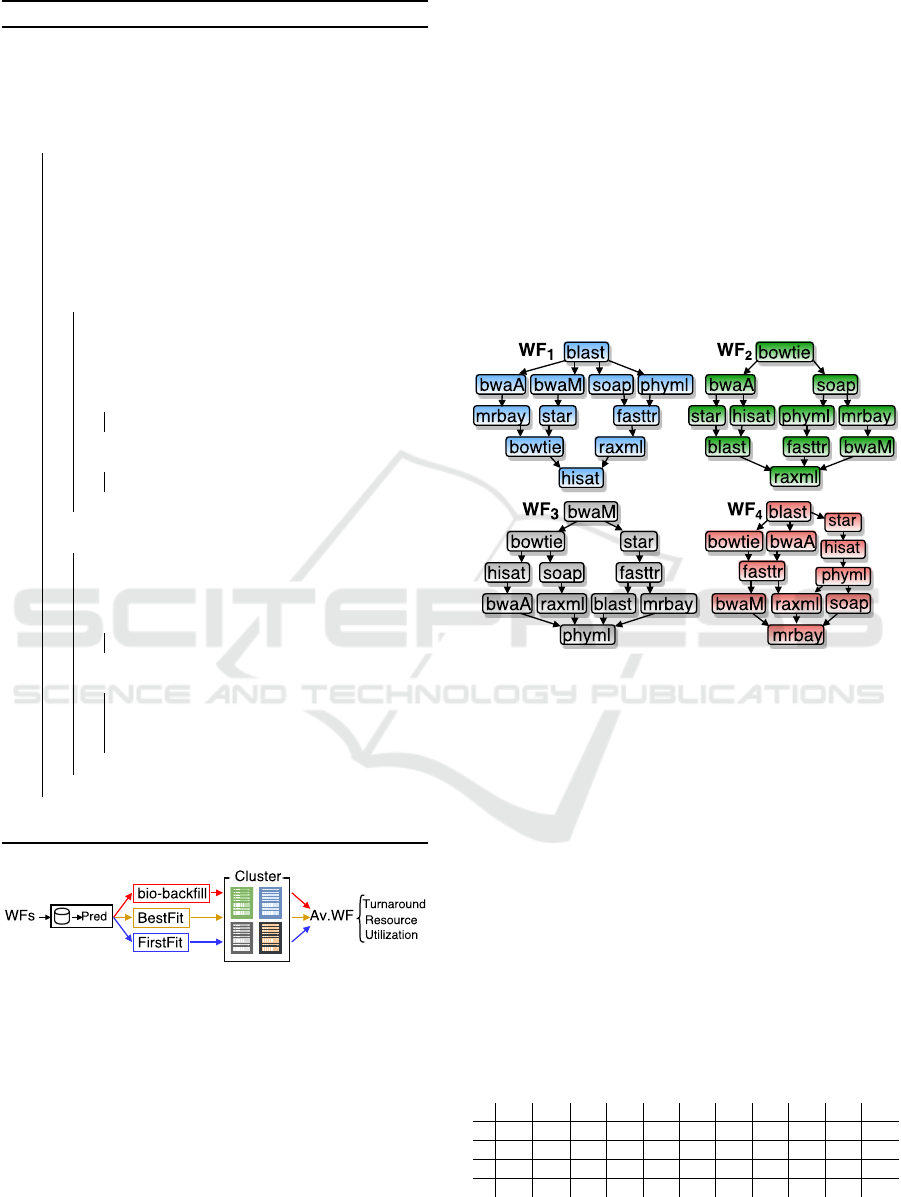
We used the 11 selected applications to build 4
workflows of the same size and arranged in different
ways. The structures of each synthetic workflow is
depicted in Figure 3. For each workflow, the same
Figure 3: Structures and applications of the 4 workflows
employed for assessing the proposal.
applications have been given different data and pa-
rameters values. Consequently, identical applications
will have different performance in each of the 4 work-
flows. Next, we analyzed the performance of work-
flows applications by executing each application in-
dependently in the cluster partition that we have at our
disposal, formed by heterogeneous nodes: 1 AMD
IO-6376 (2.3GHz, 64PU, 128GB), 1 Intel Xeon E5-
4620 (2.2GHz, 64PU, 128GB) and 2 Intel Xeon E5-
2620 (2.1GHz, 24PU, 64GB).
Although each workflow application has been exe-
cuted with different resources (AllNodes, RangePUs),
for simplicity, only average times obtained in all clus-
ter nodes with PU
MaxSpeed
, are shown in Table 1.
Table 1: Average makespans of workflows applications with
PU
MaxSpeed
, in seconds.
wf blast bwM bowt bwA hisat star soap phy mrba fastt raxm
w
1
4851 4136 3464 4391 2001 5527 5183 3719 3264 3262 3032
w
2
3760 4688 4866 4947 4145 4338 3779 3750 3890 3266 3686
w
3
4814 4547 4518 4099 4749 3359 4926 2368 3551 4554 3524
w
4
4525 5068 4871 4239 4136 4818 3445 3272 3510 4462 3309
COMPLEXIS 2018 - 3rd International Conference on Complexity, Future Information Systems and Risk
154
4.2 Proposal Assessment
The performance of the bio-backfill scheduling pol-
icy is tested, and compared with that of state-of-the-
art backfill policies: Bestfit and Firstfit. To do so,
we used WorkflowSim (Chen and E-Deelman, 2012),
an open-source tool to simulate workflows defined
with XML files, which includes various scheduling
policies. Such tool has been employed to test pre-
vious workflow scheduling algorithms such as CPFL
(Acevedo et al., 2017). The size and specifications of
the simulated cluster resources have been adjusted as
shown in Table 2. They are identical to those of our
real cluster, in which we obtained Table 1 informa-
tion.
Table 2: Specifications of the simulated cluster.
Simulator Specs
Nodes p/Cluster 4
Processors p/Node 45
Processors Freq. 6000 MIPS
RAM p/Node 96 GB
Disk capacity p/Node 1 TB
Net latency 0.2 ms
Internal latency 0.05 ms
In all the cases, all workflows are simultaneously
submitted as batch jobs, and the turnaround starts be-
ing considered until its completion. Once workflows
are submitted, the 3 algorithms proceed differently.
Firstfit and Bestfit, backfill jobs following their re-
spective criteria regarding which other high-priority
jobs must not be delayed. When doing so, multiple
jobs share the same nodes (DP>1). However, the
compatibility among jobs’ that are to share the same
nodes is not considered as a parameter in order to se-
lect the backfill candidate, leading to higher resource
competition and larger slowdown times. Conversely,
the bio-backfill policy selects the candidate consider-
ing the nodes’ loads, minimizing the resulting slow-
down. The bio-backfill proposal also includes the
WaitDep parameter to select the backfill candidate.
WaitDep allows for the scheduler to wait for non-
ready jobs that are highly compatible with the current
load to become ready and backfill them, if doing so
generates less turnaround than backfilling other, non-
compatible yet-ready jobs. Results obtained after pro-
cessing the workflows with the 3 different backfill ap-
proaches are provided in Table 3.
Results of Table 3 show how the proposed Bio-
backfill scheduler, by including the slowdown (DP
>1) and near-future dependency resolution (Wait-
Dep) as parameters to choose which candidates are
backfilled, can achieve 10% workflow turnaround re-
duction compared to Firstfit, and 7,3% compared to
Table 3: Turnarounds in seconds obtained after processing
the workflows with different backfill policies. Average im-
provement of the bio-backfill versus Firstfit and Bestfit.
BioBackfill Firstfit Bestfit Av.Improv.
WF
1
27899 29229 30248 6,19%
WF
2
30584 32407 38864 3,33%
WF
3
29211 33642 32232 11,31%
WF
4
28388 33642 31895 13,37%
Av. 29020 32230 31060 8,55%
Bestfit. Hence, the proposal improves the turnaround
of both state-of-the art backfill policies, by 8,55%, on
average. Similarly, the resource utilizations carried
out by the synthetic workflows after being processed
with the three backfill techniques, are provided in Ta-
ble 4. As mentioned in Section 3.2, the bio-backfill
scheduler algorithm uses multiple performance pre-
dictions to calculate PU
MaxSpeed
or time penalties as-
sociated with PU
LowSpeed
, and can determine whether
to allocate AvailRes or MaxRes for better resource
utilization. With that functionality, the bio-backfill
scheduler enhances resource utilization by 4,6% and
1,6% compared to Firstfit and Bestfit respectively, av-
eraging 3,8%.
Table 4: Resource Utilization of the workflows with the 3
backfill policies. Average improvement of the bio-backfill
versus Firstfit and Bestfit.
BioBackfill Firsffit Bestfit Av.Improv.
WF
1
94,6% 68,8% 85,6% 22,5%
WF
2
82,6% 69,5% 73% 16%
WF
3
74,1% 96% 89% -20%
WF
4
90,4% 88,9% 87% 2,6%
Av. 85,4% 80,8% 83,8% 3,8%
5 CONCLUSIONS
Current backfill policies improve turnaround and re-
source utilization of jobs sharing clusters, compared
to classical policies. However, they aren’t adapted for
scheduling bioinformatics workflows applications.
Hence, we developed the bio-backfill scheduler. The
scheduler includes a predictor that automatically gen-
erates performance predictions for each job with dif-
ferent resources, and the slowdowns when the DP>1.
We also developed a scheduling algorithm that back-
fills jobs, not only upon its width or length but also
by scheduling them for same-node execution so slow-
down is minimized. The algorithm includes the func-
tionality of looking ahead on the future, determining
in which cases it’s beneficial for performance to wait
for: load-compatible queued jobs to become ready,
or resources to be released. Finally, we tested the bio-
backfill on a series of bioinformatics workflows. Sim-
Bio-backfill: A Scheduling Policy Enhancing the Performance of Bioinformatics Workflows in Shared Clusters
155

ulation results show the bio-backfill proposal can im-
prove average workflow turnaround by 8,6% and re-
source utilization by 3,8% compared to state-of-the-
art Firstfit and Bestfit backfill. Present experiments
show the promising performance improvements when
adapting backfill policies to the needs of bioinformat-
ics workflows applications, proving the viability of
the bio-backfill scheduler. To further develop and test
the proposal we are currently working on applying
it into larger environments, with a greater amount of
nodes, PUs, and workflows. Future steps also include
increasing the set of applications, as well as extending
comparisons to other backfill policies.
REFERENCES
Acevedo, C., Hern
´
andez, P., Espinosa, A., and M
´
endez,
V. (2017). A Critical Path File Location (CPFL) al-
gorithm for data-aware multiworkflow scheduling on
HPC clusters. In Future Generation Computer Sys-
tems. Elsevier.
Al-Ali, R., Kathiresan, N., Anbari, M. E., Schendel, E.,
and Zaid, T. (2016). Workflow optimization of per-
formance and quality of service for bioinformatics ap-
plication in high performance computing. In Journal
of Computational Science. Elsevier.
Altschul, S., Gish, W., Miller, W., Myers, E., and Lipman,
D. (1990). Basic Local Alignment Search Tool. In
Journal of molecular biology. Elsevier.
Arabnejad, V., Bubendorfer, K., and Ng, B. (2017). Dead-
line Constrained Scientific Workflow Scheduling on
Dynamically Provisioned Cloud Resources. In Future
Generation Computer Systems, special issue. Elsevier.
Badosa, F., Acevedo, C., Espinosa, A., Vera, G., and Ripoll,
A. (2017). A Resource Manager for Maximizing the
Performance of Bioinformatics Workflows in Shared
Clusters. In International Conference on Algorithms
and Architectures for Parallel Processing. Springer.
Bucur, A. and Epema, D. (2001). The influence of com-
munication on the performance of co-allocation. In
Job Scheduling Strategies for Parallel Processing.
Springer.
Burrows, M. and Wheeler, D. (1994). A block-sorting loss-
less data compression algorithm. Citeseer.
Chen, W. and E-Deelman (2012). Workflowsim: A toolkit
for simulating scientific workflows in distributed en-
vironments. In 8th International Conference on E-
Science. IEEE.
Dobin, A., Davis, C., Schlesinger, F., Drenkow, J., Zaleski,
C., Jha, S., Batut, P., Chaisson, M., and Gingeras, T.
(2013). STAR: ultrafast universal RNA-seq aligner.
In Bioinformatics. Oxford University Press.
Figueira, S. and Berman, F. (2001). A slowdown model
for applications executing on time-shared clusters of
workstations. In Transactions on Parallel and Dis-
tributed Systems. IEEE.
Guindon, S. and Gascuel, O. (2003). A simple, fast, and
accurate algorithm to estimate large phylogenies by
maximum likelihood. In Systematic biology. Society
of Systematic Zoology.
Hatem, A., Bozda
˘
g, D., Toland, A., and C¸ ataly
¨
urek, U.
(2013). Benchmarking short sequence mapping tools.
In BMC Bioinformatics. BioMed Central.
Huelsenbeck, J. and Ronquist, F. (2001). MRBAYES:
Bayesian inference of phylogenetic trees. In Bioin-
formatics. Oxford University Press.
Kathiresan, N., Temanni, M., and Al-Ali, R. (2014). Perfor-
mance improvement of BWA MEM algorithm using
data-parallel with concurrent parallelization.
Kim, D., Langmead, B., and Salzberg, S. (2015). HISAT:
a fast spliced aligner with low memory requirements.
In Nature methods. Nature Research.
Langmead, B. (2009). Ultrafast and memory-efficient align-
ment of short DNA sequences to the human genome.
In Journal of Genome Biology.
Lawson, B. and Smirni, E. (2002). Multiple-queue back-
filling scheduling with priorities and reservations for
parallel systems. In ACM SIGMETRICS Performance
Evaluation Review. ACM.
Li, H. and Durbin, R. (2009). Fast and accurate short
read alignment with Burrows-Wheeler Transform. In
Bioinformatics. Oxford University Press.
Li, R., Li, Y., Kristiansen, K., and Wang, J. (2008). SOAP:
short oligonucleotide alignment program. In Bioinfor-
matics. Oxford University Press.
Lord, E., Diallo, A., and Makarenkov, V. (2015). Classi-
fication of bioinformatics workflows using weighted
versions of partitioning and hierarchical clustering al-
gorithms. In BMC Bioinformatics. BioMed Central.
Needleman, S. and Wunsch, C. (1970). A general method
applicable to the search for similarities in the amino
acid sequence of two proteins. In Journal of Molecu-
lar Biology. Elsevier.
NHGRI (2017). DNA Sequencing Costs: Data.
https://www.genome.gov/sequencingcostsdata/.
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). Fast-
Tree: computing large minimum evolution trees with
profiles instead of a distance matrix. In Molecular bi-
ology and evolution. Oxford University Press.
Stamatakis, A., Ludwig, T., and Meier, H. (2004). RAxML-
III: a fast program for maximum likelihood-based in-
ference of large phylogenetic trees. In Bioinformatics.
Oxford University Press.
Talby, D. and Feitelson, D. (1999). Supporting priorities
and improving utilization of the IBM SP scheduler us-
ing slack-based backfilling. In 13th International and
10th Symposium on Parallel and Distributed Process-
ing. IEEE.
Waidyasooriya, H., Hariyama, M., and Kameyama, M.
(2014). FPGA-accelerator for DNA sequence align-
ment based on an efficient data-dependent memory ac-
cess scheme. In Proceedings of the 5th International
Symposium on Highly-Efficient Accelerators and Re-
configurable Technologies.
Wu, F., Wu, Q., and Tan, Y. (2015). Workflow scheduling
in cloud: a survey. In The Journal of Supercomputing.
Springer.
Xin, H., Lee, D., Hormozdiari, F., Yedkar, S., Mutlu, O.,
and Alkan, C. (2013). Accelerating read mapping with
FastHASH. In BMC Genomics. BioMed Central.
COMPLEXIS 2018 - 3rd International Conference on Complexity, Future Information Systems and Risk
156
