
Towards an Agile Development Model for Certifiable Medical Device
Software
Taking Advantage of the Medical Device Regulation
Manuel Zamith and Gil Gonc¸alves
Faculty of Engineering of the University of Porto, Porto, Portugal
Keywords:
Medical Device, Healthcare, Agile, Software Life Cycle Models, Software Engineering, Scrum, Medical
Device Regulation, Medical Device Framework.
Abstract:
Regulation of medical devices has been one of the most prominent initiatives of the European Union in the
health domain. The recent Medical Device Regulation 2017/745/EEC extended the definition of medical
devices to standalone software systems with prognostic and prevision intended purposes. This paradigm shift
stimulates the development of more lightweight software systems such as mobile applications, that can be
classified as legitimate medical devices and can be prescribed to patients. This new context creates the need
for an urgent adjustment of the currently used software development life cycle models and processes. This
article discusses a tailored agile approach based on the Scrum model, designed to be compliant with the
international standards for medical device software development and benefit the creation of software solutions
according to the current Medical Device Framework. The discussion in this paper demonstrates there is no
reason to believe that agile methodologies should not benefit the process of creating software solutions in the
medical device domain.
1 INTRODUCTION
The strong presence of software systems in the me-
dical industry is undeniable. In fact, over 50% of all
medical devices directly depend on software in some
way (Allen, 2014). This dependency exists because
software solutions show great potential to provide a
positive change for patients, their families and even
healthcare professionals. Proof of this growth is the
fact that the medical technology ecosystem registered
more that one hundred thousand patents in the year of
2012, turning out to be the most innovative industry
in the world (Gloger, 2013).
On the other hand, this industry presents unique
challenges for software system development. The
current state of software systems in healthcare is con-
sidered to be relatively primitive (Goldsmith, 2006;
Kruger and Kruger, 2012) when compared to the fi-
nancial or retail industries. This is a direct result of
the level of complexity and demanding nature of me-
dical services.
It is impossible to create a standardized inventory
for healthcare services, rather, they must be custo-
mized at point of care, as soon as possible and ac-
cording to specific circumstances. There is also tre-
mendous variation in physician response to diagnos-
tic uncertainty from case to case, meaning that there
is great potential for collision between stakeholders at
the point of service.
Furthermore, the medical industry as a whole is
characterized by a strong influence of regulatory en-
tities concerning the development of medical devices
and information technology systems. Technological
endeavors must submit to a strict certification pro-
cess and compliance testing to a pre-established set of
standards and requirements. Compliance authorities
are motivated by the need to avoid additional risk for
patients and healthcare professionals. As such, to sur-
vive the certification processes and obtain approval, it
is certainly necessary to have high quality standards,
namely in the engineering life cycle itself.
It is paramount to reinforce that regulatory super-
vision does not concern itself only with the product’s
performance, but also with the way it’s developed.
There is a strong conviction that a rigorous, systema-
tic engineering process leads to trustworthy devices.
The idea that the products we buy and use in our
day-to-day lives should be regulated in order to ensure
that they are safe and fit for its claimed purpose is so-
mething that most individuals assumes is true. When
132
Zamith, M. and Gonçalves, G.
Towards an Agile Development Model for Certifiable Medical Device Software - Taking Advantage of the Medical Device Regulation.
DOI: 10.5220/0006861301320140
In Proceedings of the 13th International Conference on Software Technologies (ICSOFT 2018), pages 132-140
ISBN: 978-989-758-320-9
Copyright © 2018 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

we purchase furniture or articles of clothing, it’s safe
to say that we take for granted that they meet a set of
requirements guaranteeing the safety of its use. With
the devices involved in healthcare (including software
products), the need for safety is particularly impor-
tant. From a patient’s perspective, there is an assump-
tion that all systems meet a thorough set of quality
standards; and from the perspective of the healthcare
professionals, it’s imperative that quality is assured,
in order to avoid unwanted incidents that could put
their professional duties at stake (Quinn, 2017).
This paper will focus on the software engineering
development life cycle and processes that aim to make
the development of medical device software systems
more agile and at the same time guarantee their qua-
lity.
Chapter 2 will look at the entire ecosystem of le-
gal restrictions and international standards that are re-
sponsible for regulating medical device development
in the European Union. This chapter aims to give
a broad insight to what are the demands and requi-
rements software teams have to comply in order to
provide all the quality evidences needed to introduce
certified medical devices in productive environments.
Also, it will discuss an exciting change in the Euro-
pean framework that will have a great impact on the
medical device software landscape.
In chapter 3, we will discuss software develop-
ment life cycle methodologies. A comparison will be
made between plan driven and agile approaches, de-
tailing each one’s main advantages and pitfalls con-
cerning development for medical device software fol-
lowing the aforementioned requirements and stan-
dards.
As for chapter 4, it will present a novel tailored
agile process life cycle model specifically designed
to be compliant with the main standards concerning
development for medical devices.
Most definitions of critical systems base themsel-
ves on the degree of consequences that result from
a failure (Knight, 2002). According to this standard,
it’s intuitive to state that the systems developed for the
medical industry can be classified as critical.
2 MEDICAL DEVICE
DEVELOPMENT IN THE EU
Until the late nineties, each member state of the Eu-
ropean Union would implement its own approach to
regulate medical devices. However, to promote EU’s
internal market circulation, as well as to regulate an
increasingly complex market, the European Council
introduced new regulations that became known as the
”new approach directives”. These would enforce a set
of essential requirements to assure safety and perfor-
mance among medical devices (Sorenson and Drum-
mond, 2014). The directives apply to all member sta-
tes, meaning that if a medical device receives EU’s
seal of approval, it’s certified for all countries.
2.1 The Medical Device Framework
This structure, assembled by the EU to promote a
single circulation market for medical devices is cal-
led the Medical Device Framework (MDF). Up until
2017, this structure was composed by three different
directives concerning medical devices:
1. Council Directive 90/385/EEC on active implan-
table medical devices (90/385/EEC, 2007);
2. Council Directive 93/42/EEC concerning medical
devices (93/42/EEC, 2007);
3. Directive 98/79/EC of the European Parliament
and of the Council on in vitro diagnostic medical
devices (98/79/EC, 2007);
These three directives were amended by
2007/47/EC Directive in 2007 (2007/47/EC, 2007).
This framework aimed to unify the basic require-
ments for safety regarding medical devices, as well as
to specify procedures regarding documentation and
audit processes which manufacturers need to comply
with in order to produce and sell medical devices.
It is mandatory to comply with these procedures to
gain the European Council certification, as stated in
Article 4 of directive 93/42/EEC.
Medical device approval inside the EU is super-
vised by what is called a competent authority, such
as the Medicine and Healthcare Products Regulatory
Agency in the UK and INFARMED in Portugal (Kra-
mer et al., 2012). Low risk devices are declared di-
rectly to the competent authorities, that can initiate
inspections and confirm production and development
standards, as well as perform reviews on technical do-
cumentation. As for more complex devices, approval
is the responsibility of independent corporations spe-
cialized in the matter, designated by the competent
authorities (Quinn, 2017; Kramer et al., 2012).
The risk associated with the use of the medical
device is the determining factor for its classification.
The needed evidences and requirements to gain regu-
latory approval are dependent on this risk classifica-
tion. Low risk devices are classified as Class I, me-
dium risk as Class II and high risk as Class III.
The decision to be compliant with the set of requi-
rements stated in the MDF represent a clear commit-
ment for medical device manufacturers, as it demands
a significant amount of financial and time resources.
Towards an Agile Development Model for Certifiable Medical Device Software - Taking Advantage of the Medical Device Regulation
133

This can be a worthy investment because the certifi-
cation allows access to a huge market. However, be-
cause the process of acquiring the certification is such
a burden, the MDF includes the important concept of
intended purpose into the definition of what actually
constitutes a medical device. What this means is that
without the explicit intention of a manufacturer to cre-
ate a medical device, there is not one, even if the de-
vice seemingly meets every other criteria.
2.2 International Standards
In case a device manufacturer takes on the commit-
ment of being certified according to the European re-
quirements, it must behave according to a set of in-
ternational standards that supply the needed guideli-
nes to achieve it. In the healthcare domain, the ISO
13485:2016 (ISO 13485:2016, 2016) standard stipu-
lates the necessary requirements concerning quality
management systems (QMS). This standard is based
on the ISO 9001:2015 (ISO 9001:2015, 2015) and can
be considered an extension of the same. Corporations
should use these standards to make a self-evaluation
about their ability to respond effectively to their cus-
tomers’ needs.
ISO 13485:2016 does not, however, supply gui-
delines specifically for software development. The
IEC 62304:2006 standard (IEC 62304 :2006, 2006)
fills this flaw, offering the necessary recommendati-
ons concerning software development life cycles, se-
curity and maintenance of software systems for medi-
cal devices. This standard has an underlying assump-
tion that the corporation develops according to a qua-
lity management system, but does not demand certifi-
cation by the ISO 13485:2016 standard. As such, IEC
62304:2006 can be regarded as a supplement to ISO
13485:2016, specific for software development.
The IEC 62304:2006 standard bases itself on
ISO/IEC 12207:1995 (ISO/IEC 12207:1995, 1995),
that while being a comprehensive standard about soft-
ware development life cycles and processes, has al-
ready been deprecated, firstly when it was replaced
by ISO/IEC 12207:2008 and secondly when it was
upgraded into ISO/IEC 12207:2017, the most recent
version. However, the same standard is still conside-
red critical for medical device software developers as
it’s the only standard that supplies specific guidelines
for this purpose. Additionally, the norm includes a
safety classification that complements MDF’s classi-
fication. The classes shall be assigned according to
the possible effects on the patient, operator, or other
people resulting from a hazard to which the system
can contribute:
1. Class A applies in case no injury or damage is
possible;
2. Class B applies in case a non-serious injury is pos-
sible;
3. Class C applies in case death or a serious injury is
possible;
There is an obvious relationship between this clas-
sification and the one provided by the MDF. Nonet-
heless, the connection between the two is not straig-
htforward, because meanwhile MDF’s classification
applies to the device as a whole, IEC 62304:2006’s
classification only applies to software components,
which may or may not represent all the components
of the medical device, in fact, the device may include
several software components, each one with a diffe-
rent safety classification. On the other hand, if a gi-
ven software component is responsible for a critical
functionality, this link is direct, simply because a fai-
lure in this component will result in a failure of the
device. Generally, it’s safe to state that the software’s
safety classification will never be higher that the clas-
sification of the device (Pikkarainen, 2016).
The IEC 62304:2006 also provides important gui-
delines concerning the software development life cy-
cle, namely important activities that the development
model shall include.
1. The development process is of particular interest
concerning this paper. The standard demands
the creation of a development planning document,
where the manufacturer shall state all the activi-
ties involved in the engineering process, as well
as the system’s safety classification.
2. The maintenance process, where the manufactu-
rer must state what are the concrete actions that
are consequence of customer feedback.
3. The risk management process. The manufacturer
shall document a process for risk management and
include it in the development plan. For such, the
14971:2007 shall be used.
4. The configuration management process, must
also be included in the development plan and
includes a list of all items that must be controlled,
traced and versioned.
5. The problem resolution process. The standard
dictates that the manufacturer must provide a re-
port for each identified problem, as well as detail
strategies for investigating and resolving the pro-
blem.
Even with this set of recommendations, it’s im-
portant to reinforce all the liberties given by the stan-
dard. It does not make restrictions concerning the
contents of the produced documentation and, most
ICSOFT 2018 - 13th International Conference on Software Technologies
134

importantly, it does not enforce a development life cy-
cle model.
2.3 The Medical Device Regulation
The IEC 62304:2006 makes the assumption that all
software components are part of an embedded medi-
cal device and, as a result, it does not include all the
system requirements for software. The standard that
aims to achieve this is IEC 60601-1 (IEC 60601-1-
11:2015, 2015), concerning electrical equipment, ho-
wever, in 2007, when MDF’s directives where amen-
ded, standalone software systems where included in
the definition of medical device, meaning that soft-
ware products could be officially classified as medical
devices.
This amendment unveiled a gap in the internati-
onal standards to date, because of the fact that there
was no publication with guidelines concerning stan-
dalone behavior of software as legitimate a medical
device (McHugh et al., 2011). The 2007/47/EC di-
rective includes software in its definition of medical
device and goes beyond that, reinforcing this idea in
Recital 6:
It is necessary to clarify that software in its
own right, when specifically intended by the
manufacturer to be used for one or more of
the medical purposes set out in the definition
of a medical device, is a medical device. Soft-
ware for general purposes when used in a he-
althcare setting is not a medical device.
The profound changes introduced by this amend-
ment gave wings to the possibility of creation of stan-
dalone software certified as a medical device, which
is the case of FibriCheck, a smartphone application
for the monitoring of arrhythmias(FibriCheck, ).
As of April 2017, the Regulation for Medical De-
vices 2017/45/EC (RDM) (2017/745/EEC, 2017) was
adopted by the European Union, and replaced MDF’s
directives 90/385/EEC and 93/42/EEC, as well as its
amendments. Unlike directives, regulations are di-
rectly applicable to all member states without the
need of an individual implementation in each mem-
ber state’s local law.
This regulations introduces significant chan-
ges to its predecessors in some aspects. Fir-
stly, the definition of medical device was ex-
panded for equipments that aim to perform
diagnostic or prediction of diseases. As such, a
software application capable of measuring the risk
for contracting some disease based on data collected
through sensors could be certified as a medical de-
vice, if the manufacturer declares this as an intended
purpose.
The regulation states exceptions for devices whose
intended purposes are related to non-clinical duties
such as lifestyle and well-being, but manufacturers
have to provide proper justification, as to avoid ar-
bitrary classifications to avoid the burden of certifica-
tion.
This expansion of the definition of medical device,
put together with new safety classification rules spe-
cifically for software has exciting implications for the
software development industry. According to the new
regulations many applications that did not fit the de-
finition of medical device now do, probably with a
classification of Class II. Any software enthusiast can
imagine the implications for corporations that work
with technologies such as big data and analytics, ex-
perts in the field of prognostic and prediction (Lee and
Yoon, 2017).
3 SOFTWARE LIFE CYCLES FOR
MEDICAL DEVICE
DEVELOPMENT
Software engineering is a big part of what makes soft-
ware products reliable and valuable. As stated by
the IEEE, it consists of the application of a systema-
tic, disciplined, quantifiable approach to the develop-
ment, operation and maintenance of software (IEEE,
1993). Additionally, software engineering processes
define the needed structure to deliver technology so-
lutions efficiently (Ian Sommerville, 2010). They pro-
vide a basis for project control and management, con-
text from which methods and techniques are applied,
milestones are defined and quality is assured.
At this stage, it’s extremely important to state that
a software engineering process does not aim to be a
rigid prescription of how to build software products,
rather, it should be faced as an adaptable approach for
teams to pick the most appropriate set of tasks and
actions according to the available resources. The ul-
timate goal should always be to deliver high quality
products in the established time frame and obtain cu-
stomer satisfaction.
Even though that’s true, life cycle models for soft-
ware development provide an important script for
the success of any project (Pressman, 2009) and the
choice of which one to follow is always an interesting
argument to have. Some would argue that the only
irresponsible strategy would be to try to standardize
and impose the same development model for every
situation (Vogel, 2011), but the benefits of following
a model are consensual. Without following defined
steps during development, it becomes extremely hard
Towards an Agile Development Model for Certifiable Medical Device Software - Taking Advantage of the Medical Device Regulation
135

to keep track of the project’s state, manage milesto-
nes and assign tasks to team members. This control
allows an increase of productivity and overall quality,
but more importantly in the domain of critical sys-
tems, it allows to create quality evidences for regula-
tory entities.
The intangible nature of software allows for a
great variety concerning life cycle models, that can
differ in aspects such as overall flow, work products,
level of discipline, customer involvement and team
autonomy.
3.1 Plan Driven Approaches
Considered the first approach to software develop-
ment, the waterfall model, credited to Winston W.
Royce (Royce, 1970), is a popular example of a plan-
driven approach to software development. It por-
trays unidirectional communication between the cy-
cle’s activities. This model is considered to be linear,
because it suggests a sequential systematic approach.
Although originally Royce predicted a certain degree
of iteration between consecutive activities (feedback
loops), most corporations implement straightly linear
waterfall methodologies.
The process begins with a requirements specifi-
cation activity, in which the main functionality and
services provided by the application are agreed upon
with the relevant stakeholders. After that, the team
designs the system, building a global architecture
from the elicited requirements. The implementation
activity follows and the solution is built, before being
integrated and tested. There is also room for a mainte-
nance phase, that implies bugfixing and overall impro-
vement of the product. At each phase of the process,
high priority is given to documentation.
The waterfall model is still a big favorite for deve-
lopment in the healthcare industry because of its plan
driven nature (McCaffery et al., 2016). Its stringency,
attention to quality and priority to documentation sti-
mulate the production of tangible evidences needed to
satisfy regulations and audits.
On the other hand, this line of thought can be-
come less appealing if the software solution has a
more unpredictable nature concerning scope and re-
quirements. The first working version of the software
solution is only finalized in later stages of the deve-
lopment process and that can lead to very high costs
in case something has to be changed.
The strictness of plan-driven approaches is advan-
tageous in some points, mainly when certification is
needed, as we have seen. However, with the intro-
duction of the Medical Device Regulations, one can
predict a change in the nature of the software aiming
to become certified as medical devices, namely be-
cause more lightweight standalone applications will
become relevant. Using a linear model in these ca-
ses can lead to frustration and become unappealing
(Spence, 2005).
3.2 Agile Approaches
In the eighties and beginning of the nineties, linear
life cycle models based on quality assurance and strict
planning were considered the standard approach to
obtain quality. This vision came from experience de-
veloping critical systems (Ian Sommerville, 2010).
However, as this strategy began to be applied by teams
in much smaller businesses, the overhead implied by
the implementation of the development model itself
became unbearable. Most of the resources available
were needed just for quality assurance tasks, and not
for development tasks.
The consequent discontent concerning waterfall
approaches lead to the uprising of the Agile Mani-
festo (Agile Alliance, 2001). This line of thought gi-
ves high priority to ideas such as customer collabora-
tion, reaction to change and communication between
team individuals and disregards production of com-
prehensive documentation and following a rigid plan.
What makes agile so appealing is the priority gi-
ven to accommodate change. In a traditional model,
the cost of change rises non-linearly as the project
progresses. This means that at later stages, when the
stakeholders see the finished product, it’s extremely
costly to change anything. Agile proposes that change
should be possible at any stage of the development
process without major costs.
But the agile philosophy is more than that. It’s a
work paradigm where communication is encouraged
between team members and fast delivery of working
solutions is emphasized (Warden and Shore, 2008).
SCRUM is a popular implementation of the agile
philosophy introduced by Ken Schwaber and Jeff Su-
therland (Schwaber, 1997). It proposes incremental
deliveries of working products through iterative cy-
cles.
The SCRUM team includes an element called a
Product Owner, who is the person in charge of maxi-
mizing the overall value of the solution, which is de-
signed and built by the development team. He is re-
sponsible for prioritizing and ordering the develop-
ment tasks, which are called user stories, in a product
backlog. The development team should guaranty a
functional delivery at the end of each increment.
The time frame between each delivery is called a
sprint. The scrum master, who is part of the deve-
lopment team, is in charge of making sure the scrum
ICSOFT 2018 - 13th International Conference on Software Technologies
136

process is followed and understood by all of those in-
volved.
The plan-driven versus agile discussion is an inte-
resting and relevant one. Discipline is the foundation
for success at any diligence: athletes train, musicians
practice to perfect their technique and engineers refine
their processes. In the software world, craftsmans-
hip is a term starting to gain popularity and describes
the art of developing high quality, detail oriented soft-
ware. On the other hand, agility is flexible and inven-
tive, when discipline is rigid and predictable. Agility
provides ability to exit comfort zones and react to the
unexpected.
It is our strong opinion that any endeavor demands
discipline and agility to succeed. Without agility, dis-
cipline translates to strong bureaucracy and stagna-
tion; nonetheless, the opposite scenario is nothing but
a startup that has yet to think about measurable profit
(Boehm, 2004).
Over the past two decades, the software develop-
ment community has been defied by the agile mo-
vement to change its perspective radically. As afo-
rementioned, the medical device landscape is also in
the verge of drastic changes, with the introduction
of standalone, lightweight applications into its ecosy-
stem.
Although plan-driven methodologies have domi-
nated the healthcare industry in the past, there is re-
ason to argue in favor of the need to make the deve-
lopment processes more agile, in light of the medi-
cal device regulation’s appearance. The ultimate goal
should be to find a development model able to com-
ply with regulations and standards, but also provides
enough flexibility and ability to react to change.
4 A TAILORED AGILE
APPROACH
Software process tailoring is the activity of tuning a
process to meet the needs of a specific project or con-
text (Xu and Ramesh, 2008). We suggest a develop-
ment life cycle model that allows compliance with the
MDF and also IEC 62304:2006. This model is also
an adaptation of SCRUM, allowing it to be agile and
highly reactive to changes and scope. The ultimate
goal is to provide the much needed structure for de-
velopment of lightweight solutions certifiable as me-
dical devices.
4.1 Life Cycle Description
An illustration of the suggested model is presented in
figure 1. The proposed activities are characterized as
follows:
• Product Vision: Should normally happen during
project proposal (in case there is a defined cus-
tomer) and has no predetermined duration. Du-
ring the product vision phase, the product ow-
ner, scrum master and user experience designers
work together in order to create an overall vi-
sion of the product. A macro requirements analy-
sis should be performed to gain better understan-
ding of the solution, as well as general architec-
ture concepts (system design) and user interface
previews. The outcomes of this activity should be
an extensive product backlog, a software architec-
ture document and a software plan document (as
demanded by the IEC 62304:2006 standard). By
the end of the product vision, the team must have
defined plans regarding traceability, risk manage-
ment, problem resolution, testing and configura-
tion management. Standards and conventions for
development should also be defined at this stage.
• Sprint 0 (optional): Unlike the remaining sprints,
Sprint 0 does not include SCRUM’s ceremonies,
as it is not an official Scrum sprint. It is fully de-
voted to reviewing the product vision’s outcomes.
Interface specifications and end-to-end proofs of
concept should be performed in order to validate
the proposed architecture (the complete develop-
ment team should be available). Outputs of this
phase include a full review of the product backlog,
grooming of the backlog for Sprint 1, updated pro-
ject plans and development infrastructure (Conti-
nuous Integration Server, Integrated Development
Environment (IDE) configurations, test infrastruc-
ture). Another important outcome of Sprint 0 is
the definition of ”Done”, which must be under-
stood by the whole Project Team and equally ap-
plied across Sprints.
• Sprint: A Sprint is the development iteration, with
duration between two and four weeks. This du-
ration can vary according to the unique features
of the medical device. Each Sprint aims at com-
pletely developing a scope, a product increment,
meaning that it must include all required develop-
ment, integration and testing activities to have a
functional and demonstrable version of the soft-
ware product at the end of the Sprint. When a
Sprint kicks-off, elements outside the Develop-
ment Team are not allowed to interfere in that
Sprint. It is the Scrum Master’s job to protect
Towards an Agile Development Model for Certifiable Medical Device Software - Taking Advantage of the Medical Device Regulation
137
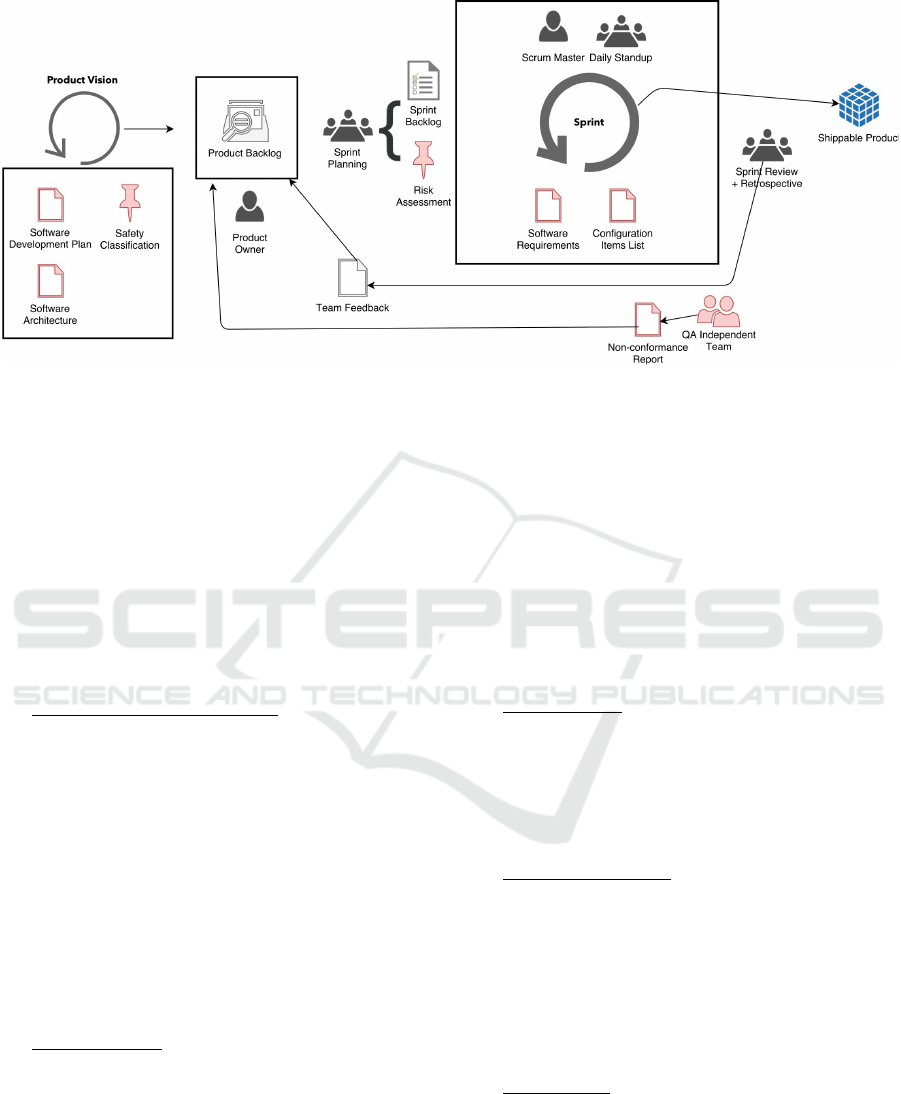
Figure 1: Tailored Scrum life cycle model. The model aims to provide a structure for compliance according to the IEC
62304:2006 standard, but also provide agile’s characteristic flexibility and capability to react to changes in scope . All items
colored in red are not contemplated in the default Scrum implementation and were introduced as tailored items.
the development team from external interferen-
ces. All change requests are solely handled by
the Scrum Master and only applicable in future
Sprints. A Sprint is ”Done” when all items are
”Done”, when all acceptance criteria are met and
Sprint Goal is achieved. As outputs of each sprint,
an updated specification of software requirements
and an updated configuration items list must be
produced in order to show evidence of these acti-
vities to regulatory authorities.
• Quality Assurance checkpoint: At the end of each
sprint, an independent quality assurance team per-
forms an internal audit, reviewing deliverables
and all evidences for certification by the medical
device regulation and the IEC 62304:2006 stan-
dard. The members of this team must not be part
of development team. A non-conformance report
should be built and non-conformities introduced
in the product backlog by the product owner. This
approach was already implemented successfully
in the R-Scrum model (Fitzgerald et al., 2013).
4.2 Scrum Ceremonies
• Sprint Planning: Each Sprint starts with a Sprint
Planning Meeting. This ceremony has the pur-
pose of defining the scope of the Sprint and de-
tailing the tasks to accomplish that scope. During
this meeting, the team must decide on the scope
to be done (What to do?), and how they will in-
clude that functionality (How to do?). The pro-
duct owner has the responsibility of presenting an
updated and prioritized product backlog, so that
the team can accurately estimate each user story
to be included in the next sprint (Sprint Backlog).
Additionally, during the sprint meeting, the pro-
duct owner and scrum master should identify the
risks concerning the items selected for the sprint.
At each planning, these risk should be reevalua-
ted and iterated. Risk management is a very im-
portant part of medical device development and
evidences of this are needed for certification. Ul-
timately, the product owner should be responsible
for managing the risk management items.
• Sprint Review: The Sprint Review Meeting serves
the purpose of collaborating on assessing the va-
lue of what was done in the Sprint and what are
the next things that could be done. This ceremony
provides valuable input to the Product Owner to
update the Product Backlog and subsequent Sprint
Planning meeting.
• Sprint Retrospective: The purpose of this meeting
is for the Team and the Scrum Master to look back
at the Sprint to make the process more effective -
delivering the user stories - and more enjoyable
- more satisfactory for the Team members. This
meeting takes place after the Sprint Review Meet-
ing and before the next Sprint Planning Meeting.
The findings of this meeting must be in the form
of actionable improvement measures.
• Daily Scrum: Daily management of Sprint execu-
tion is performed by conducting daily short meet-
ings (15 minutes maximum).
4.3 Process Validation
The proposed model will be validated by a team of
students of the Faculty of Engineering of the Univer-
ICSOFT 2018 - 13th International Conference on Software Technologies
138

sity of Porto. The students will be developing a stan-
dalone software system, provisionally classified as a
Class C medical device.
The main goal of this study will be to implement
the suggested tailored Scrum process and evaluate if
it is beneficial for the development team in two key
aspects:
1. The process allows the final product to be success-
fully classified as a medical device, according
to the Medical Device Regulations and IEC
62304:2006.
2. The process does not put Scrum and Agile’s main
advantages at jeopardy by introducing additional
overhead.
These conclusions can be obtained through exis-
ting metrics and studies. For example, Abrahamsson
et al. presented a study where software life cycles are
analyzed and evaluated (Abrahamsson et al., 2002).
Alson, Boehm (Boehm, 2004) presented a compre-
hensive comparison between different models in or-
der to study the balance between agility and disci-
pline. The author collected metrics like budget perfor-
mance, schedule achievement, team satisfaction and
defect rate.
5 CONCLUSIONS
Balancing agility and discipline is everything but a
simple task when dealing with software development
life cycles, particularly in the critical system domain,
such as is the case with medical devices.
However, there are no tangible evidences preven-
ting agile methodologies from benefiting develop-
ment teams in the task of building valuable medical
devices. In 2017, with the introduction of the new
Medical Device Regulations, one can expect the deve-
lopment of standalone software systems legally cer-
tified as medical devices which are lightweight and
operating in fast changing environments. This de-
mands a renewal of the way the software community
is used to building these systems.
It is a fact ”out of the box” that Agile methods
such as Scrum have some shortfalls regarding highly
regulated ecosystems (McHugh et al., 2012). Ho-
wever, these barriers are either a result of misunder-
standings of the agile philosophy or can be overcome
through tailoring. For example, it’s generally assu-
med that agile disregards documentation, but these
documents can be written if they are highly prioriti-
zed by the product owner (agile aims to deliver va-
lue most of all). Traceability can be achieved through
the use of the appropriate tools and risk management
evidences can be created, as we have shown, in each
sprint planning meeting.
The software community has reason to embrace
these changes in the medical device landscape with
excitement, because they represent an opportunity to
create software that can make positive changes in pe-
ople’s lives on a daily basis. Given these new circum-
stances, a mobile application can very well be certi-
fied as a medical device, and this means it can possi-
bly be prescribed to a patient by a medical doctor. On
the other hand, development processes must be ret-
hought to face these challenges.
ACKNOWLEDGEMENTS
The author would like to thank Critical Software and
it’s delivery management team for providing him the
possibility to pursue this study in the field of software
engineering.
REFERENCES
2007/47/EC (2007). Directive 2007/47/EC of the european
parliament and of the council amending Council Di-
rective 90/385/EEC on the approximation of the laws
of the Member States relating to active implantable
medical devices, Council Directive 93/42/EEC con-
cerning medical devices and Directive 98/8/EC con-
cerning the placing of biocidal products on the market.
Diretiva, European Union.
2017/745/EEC (2017). Regulation (EU) 2017/745 of the
European Parliament and of the council on medi-
cal devices, amending Directive 2001/83/EC, Re-
gulation (EC) No 178/2002 and Regulation (EC)
No 1223/2009 and repealing Council Directives
90/385/EEC and 93/42/EEC. Regulamento, European
Union.
90/385/EEC (2007). Council Directive 90/385/EEC on
active implantable medical devices. Diretiva, Euro-
pean Union.
93/42/EEC (2007). Council Directive 93/42/EEC concer-
ning medical devices. Diretiva, European Union.
98/79/EC (2007). Directive 98/79/EC of the European Par-
liament and of the Council on in vitro diagnostic me-
dical devices. Diretiva, European Union.
Abrahamsson, P., Salo, O., Ronkainen, J., and Warsta, J.
(2002). Agile software development methods: Review
and analysis. pages 3–107.
Agile Alliance (2001). What is Agile Software Develop-
ment?
Allen, S. (2014). Medical device software under the mi-
croscope. Network Security, 2014(2):11–12.
Boehm, B. (2004). Balancing Agility and Discipline: A
Guide for the Perplexed. pages 1–1.
Towards an Agile Development Model for Certifiable Medical Device Software - Taking Advantage of the Medical Device Regulation
139

FibriCheck. FibriCheck - Physicians.
Fitzgerald, B., Stol, K.-J., Sullivan, R. O. ., and O ’brien, D.
(2013). Scaling Agile Methods to Regulated Environ-
ments: An Industry Case Study. Proceedings of the
2013 International Conference on Software Engineer-
ing, pages 863–872.
Gloger, B. (2013). 3 Reasons for Introducing Agile Pro-
duct Development in Medical Technology. Technical
report.
Goldsmith, J. C. (2006). The healthcare information techno-
logy sector. In Burns, L. R., editor, The Business
of Healthcare Innovation, pages 322–347. Cambridge
University Press, Cambridge.
Ian Sommerville (2010). Software Engineering. Lecture
Notes in Computer Science. Pearson, Berlin, Heidel-
berg.
IEC 60601-1-11:2015 (2015). IEC 60601-1-11:2015. Me-
dical electrical equipment – Part 1-11: General requi-
rements for basic safety and essential performance –
Collateral standard: Requirements for medical electri-
cal equipment and medical electrical systems used in
the home healthcare environment. Standard, ISO.
IEC 62304 :2006 (2006). IEC 62304 :2006. Medical device
software – Software life cycle processes. Standard,
IEC.
IEEE (1993). EEE Standards Collection: Software Engi-
neering. In IEEE Standard 610.12-1990.
ISO 13485:2016 (2016). ISO 13485:2016. Medical devices
– Quality management systems – Requirements for re-
gulatory purposes. Standard, ISO.
ISO 9001:2015 (2015). ISO 9001:2015. Quality manage-
ment systems – Requirements. Standard, ISO.
ISO/IEC 12207:1995 (1995). ISO/IEC 12207:1995. Infor-
mation technology – Software life cycle processes.
Standard, ISO, IEC.
Knight, J. C. (2002). Safety Critical Systems: Challenges
and Directions. In Proceedings of the 24th internati-
onal conference on Software engineering - ICSE ’02,
page 547, New York, New York, USA. ACM Press.
Kramer, D. B., Xu, S., and Kesselheim, A. S. (2012). Re-
gulation of Medical Devices in the United States and
European Union. New England Journal of Medicine,
366(9):848–855.
Kruger, K. H. and Kruger, M. A. (2012). The medical
device sector. In Burns, L. R., editor, The Business
of Healthcare Innovation, pages 376–450. Cambridge
University Press, Cambridge.
Lee, C. H. and Yoon, H.-J. (2017). Medical big data: pro-
mise and challenges. Kidney Research and Clinical
Practice, 36(1):3–11.
McCaffery, F., Trektere, K., and Ozcan-Top, O. (2016).
Agile Is it Suitable for Medical Device Software De-
velopment? In Software Process Improvement and
Capability Determination, volume 770 of Communi-
cations in Computer and Information Science, pages
417–422. Springer International Publishing.
McHugh, M., McCaffery, F., and Casey, V. (2011). Standa-
lone Software as an Active Medical Device. In Com-
munications in Computer and Information Science,
volume 155 CCIS, pages 97–107.
McHugh, M., McCaffery, F., and Casey, V. (2012). Bar-
riers to Adopting Agile Practices When Developing
Medical Device Software. In Software Process Im-
provement and Capability Determination, pages 141–
147.
Pikkarainen, M. (2016). Introducing Agile Practices into
MDevSPICE
R
. 8(1):133–142.
Pressman, R. S. (2009). Software Engineering: A Practiti-
oner’s Approach. McGraw-Hill, 7th edition.
Quinn, P. (2017). The EU commission’s risky choice for a
non-risk based strategy on assessment of medical de-
vices. Computer Law & Security Review, 33(3):361–
370.
Royce, D. W. W. (1970). Managing the Development of
large Software Systems. Ieee Wescon, (August):1–9.
Schwaber, K. (1997). SCRUM Development Process.
In Business Object Design and Implementation, vo-
lume 6, pages 117–134. Springer London, London.
Sorenson, C. and Drummond, M. (2014). Improving Medi-
cal Device Regulation: The United States and Europe
in Perspective. Milbank Quarterly, 92(1):114–150.
Spence, J. (2005). There has to be a better way! [soft-
ware development]. In Agile Development Conference
(ADC’05), pages 272–278. IEEE Comput. Soc.
Vogel, D. (2011). Medical Device Software: Verification,
Validation and Compliance. Artech House.
Warden, S. and Shore, J. (2008). The Art of Agile Develop-
ment. O’Reilly Media.
Xu, P. and Ramesh, B. (2008). Using process tailoring to
manage software development challenges. IT Profes-
sional, 10(4):39–45.
ICSOFT 2018 - 13th International Conference on Software Technologies
140
