
In Silico Analysis of the Phytochemical Compounds in Carica papaya
Seeds for Optimizing the Inhibitors of HMG-CoA Reductase
Hariyanti, Rizky Arcinthya Rachmania, Mutia Karinah, and Hadi Sunaryo
Faculty of Pharmacy and Science, Universitas Muhammadiyah Prof. DR. HAMKA
Keywords: Carica papaya seed, HMG-CoA reductase, Molecular docking, Anti-hypercholesterolemia.
Abstract: HMG-CoA Reductase, a key enzyme in the cholesterol biosynthesis, catalyzes the conversion of 3-
hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) into mevalonate. Therefore, this enzyme is the target
of the cholesterol-lowering drugs known as statins. Carica papaya seed extract contains phytochemical
compounds that are thought to have a cholesterol-lowering effect. The present study was designed to
examine the ability of the secondary metabolites of Carica papaya seeds as an antagonist to HMG-CoA
reductase using in silico molecular docking. The docking analysis was carried out in PLANTS 1.2 software
in which the lowest ChemPLP score, i.e., free energy, was the molecular docking parameter. Seven ligands
were docked with HMG-CoA reductase receptor, three of which were benzyl glucosinolate, oleic acid, and
glucotropaeolin that had the best ChemPLP scores, namely -90.5491 kcal/mol, -81.7665kcal/mol, and -
85.1919 kcal/mol, respectively. Benzyl glucosinolate formed hydrogen bonds with the active site of the
targeted protein. As a conclusion, this compound can inhibit the enzyme HMG-CoA reductase, and it has
the potential for anti-hypercholesterolemia
.
1 INTRODUCTION
Hypercholesterolemia, excessively high levels of
plasma cholesterol, emerges as a strong risk factor
for cardiovascular disease (CVD) (Stapleton et al.,
2010). Cholesterol is an important component of the
cell membrane and is essential for the synthesis of
various important metabolites. HMG-CoA
Reductase (HMGCR), a key enzyme in the
cholesterol biosynthesis, catalyzes the conversion of
3-hydroxy-3-methylglutaryl coenzyme A (HMG-
CoA) into mevalonate. Human HMGCR consists of
polypeptide chains of 888 amino acids with three
functional portions: residues 1-339 span the
membrane of the endoplasmic reticulum eight times,
while residues 340-459 connect the membrane
portion to the catalytic portion (i.e., residues 460-
888), which resides in the cytoplasm. This enzyme is
anchored in the membrane of the endoplasmic
reticulum, which has seven transmembrane domains,
with the active site located in a long carboxyl-
terminal domain in the cytosol (Nakanishi et al.,
1988). The inhibition of this enzyme results in a
significant decrease in cholesterol levels (Goldstein
and Brown, 1990).
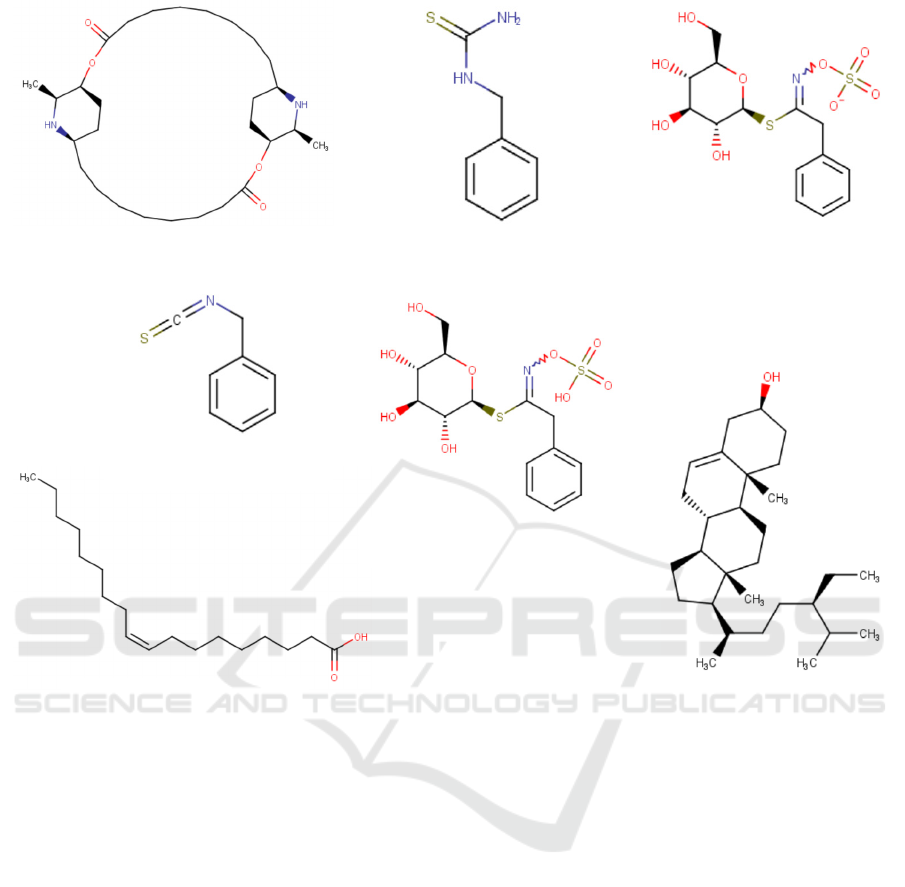
Carica papaya seeds contain some compounds
that are suspected to have a cholesterol-lowering
effect on the mechanism of inhibiting the enzyme
HMG-CoA reductase. The phytochemical
substances in Carica papaya seeds have been
reported to contain flavonoids, saponins, and tannins
(Olivera et al., 2007). These compounds can
decrease the HMG-CoA reductase activity and,
therefore, inhibit cholesterol synthesis (Siregar
2015; Afrose et al., 2010). The main components of
papaya seeds are fatty acids, crude protein, crude
fiber, papaya oil, carpaine, benzyl isothiocyanate,
benzyl glucosinolate, glucotropaeolin, benzyl
thiourea, hentriacontane, β-sitosterol, caricin, and
myrosin enzyme (Yogiraj et al., 2014). Also, there
are other compounds, such as alkaloids, steroids,
essential oils, oleic acid, and palmitic acid (Satriyasa
and Pangkahila, 2010). Oleic acid is part of the fatty
acids found in papaya seeds. Natali et al. (2007)
state that oleic acid has an inhibitory effect on
HMG-CoA reductase enzyme.
The mechanism of interaction between HMG-
CoA reductase enzyme and the compounds in
papaya seed (Figure 1) can be investigated using
molecular docking method. Molecular docking is
used to discover compounds for potentially potent
Hariyanti, ., Rachmania, R., Karinah, M. and Sunaryo, H.
In Silico Analysis of the Phytochemical Compounds in Carica papaya Seeds for Optimizing the Inhibitors of HMG-CoA Reductase.
DOI: 10.5220/0008240501230132
In Proceedings of the 1st Muhammadiyah International Conference on Health and Pharmaceutical Development (MICH-PhD 2018), pages 123-132
ISBN: 978-989-758-349-0
Copyright
c
2021 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
123
drugs in relatively short periods of time (Zukrullah
et al., 2012). Based on this background, this research
attempted to determine the mechanism of the
interaction of HMG-CoA reductase enzyme and the
ligands of the compounds in papaya seeds. The
enzyme was rolled with each ligand in PLANTS 1.2
software and then visualized to see the interaction
formed between the ligand and the receptor in
Molecular Molegro Viewer (MMV) software. Using
the Lipinski Rule of Five, the in silico analysis
determined any compounds that had oral
bioavailability.
2 MATERIALS AND METHODS
2.1 Materials
The molecular docking program was run in LINUX
with UBUNTU 16 system (64 bit). The ligand
design and visualization were developed using
Windows 10 operating system. The software used in
this research included PLANTS 1.2
(http://www.tcd.unikonstanz.de/ research/plants.php)
for docking, YASARA 17.4.17 (http:
//www.yasara.org/viewdl.htm) for protein
preparation and visualization, Marvinsketch 17.9.0
(http://www.chemaxon.com/marvin/download-
user.html) for ligand preparation, and Molegro
Molecular Viewer 2.5 for visualization.
The 3-dimensional crystallographic structure of
HMG-CoA reductase was downloaded from the
Protein Data Bank at http://www.rcsb.org/pdb in
.pdb format (Purnomo, 2013). Previously, this
research had consulted scientific journals to
determine the receptor. The 3D structures of the
ligands included simvastatin acid and simvastatin, as
well as the phytochemical compounds from Carica
papaya seeds, i.e., carpaine, benzyl isothiocyanate,
benzyl glucosinolate, glucotropaeolin, benzyl
Figure 1: The ligand structures of the phytochemical compounds in Carica papaya seeds
Carpaine
Glucotropaeolin
Oleic Aci
d
β-Sitosterol
Benzyl Thiourea
Benzyl Isothiocyanate
Benzyl Glucosinolate
MICH-PhD 2018 - 1st Muhammadiyah International Conference on Health and Pharmaceutical Development
124

thiourea, β-sitosterol, and oleic acid. These
structures were designed in Marvin Sketch in .mol2
and .mrv formats
.
2.2 Methods
2.2.1 Protein Preparation
The protein macromolecule of HMG-CoA reductase
with the PDB code 1HW9 for homo sapiens
downloaded from the Protein Data Bank at
http://www.rcsb.org/ was inserted into YASARA
software for preparation. The protein
macromolecules were separated from solvents and
ligands or non-standard residues. The separation of
macromolecules from unnecessary molecules used
YASARA program (edit > delete > residue). The
elimination of water molecules (edit > delete >
water) and the addition of hydrogen to the structures
(edit > add > hydrogen to all) were also run in this
program. The results were stored with the protein
name and in .mol2 format.
2.2.2 Ligand Preparation
The ligand structures were downloaded from
www.pubchem.ncbi.nlm.nih.gov in 2D model. The
protonation was changed at pH 7.4 using the Marvin
Sketch (Calculation > Protonation > Major
Microspecies), the resulted data were then stored in
.mrv files. These files were opened, and a
conformational search in the same software was
stored in .mol2 format (Calculation > Conformation
> Conformer).
2.2.3 Validation of Molecular Docking
Methods
Before the virtual screening, validation was
performed to determine the values of the root mean
square distance (RMSD). It was run in the YASARA
program (Analyze > RMSD > Molecule) by entering
specific ligands and receptors in .mol2 format. A
protocol was accepted if the RMSD of the heavy
atom was smaller than 2.0 Å.
2.2.4 Molecular Docking with PLANTS 1.2
Software
The molecular docking was processed in PLANTS
program. This software can only run in Linux
operating system. All data and PLANTS
applications were moved from the desktop to the
root (sudo -s), and the terminal was opened in Linux
afterward, The command "cp
/home/desktop/PLANTS1.2 PLANTS" was typed in
and followed by the command "chmod u + x
PLANTS" to activate the PLANTS application. The
results of the ligand and receptor preparations were
stored in .mol2 and moved to the root with the
command "cp /home/desktop/*.mol2."
The next step was to find the binding site using
the command "./PLANTS - bind ref_ligand.mol2 5
protein.mol2". To examine whether the settings on
the PLANTS were correct, this research used the
command "kwrite plantsconfig", followed by
"./PLANTS --mode screen plantsconfig". When the
docking process was complete, the results were
displayed in the terminal by entering the command
"cd results /" and, then, "more bestranking.csv".
From the ten docking results, the one with smaller
conformation value was selected, and the result was
stored using the command "cp*_entry_
(conformation number)_conf_01.mol2/home/
desktop/".
2.2.5 Molecular Docking Result Analysis
And Visualization
The docking results were observed from the output
in a notepad format. The complex conformation of
the docking result was determined by choosing
conformation based on the CHEMPLP score, i.e.,
the lowest free energy. The docking results were
visualized using YASARA software to determine
whether the hydrogen bond distance was <3.5 Å.
2.2.6 Drug Scan Analysis
The drug scan was analyzed on a website
(http://chemicalize.com). The analysis involved
uploading the ligand file in .mol format. Then, the
results were downloaded in PDF format.
3 RESULTS AND DISCUSSION
The initial stage of the docking process was the
preparation of the protein structure where the
proteins were selected from the GDP site
purposively. The HMG-CoA reductase enzyme in
.pdb format was downloaded from the protein
database organized by the Research Collaboratory
for Structural Bioinformatics (RCSB) at
http://www.rcsb.org/. The protein chosen for the
HMG-CoA reductase was 1HW9 (PDB code). It is
an enzyme complex with simvastatin acid
(endogenous ligand). The protein structure
In Silico Analysis of the Phytochemical Compounds in Carica papaya Seeds for Optimizing the Inhibitors of HMG-CoA Reductase
125

downloaded from GDP generally still contains a
solvent (water). In this study, the protein structures
of the other residues were analyzed without the
endogenous ligand structure. In other words, the
protein structures depicted a protein without
endogenous ligand and other molecules such as
water and other single atoms; therefore, the docking
process only analyzed the interaction of test
compounds and proteins (Kitchen et al., 2004).
These water ligands and molecules had to be
removed from the protein macromolecules as they
might prolong the duration of the docking
simulation. The addition of the hydrogen atom in
question afterward aimed to bring up the existing
hydrogen atoms in the structure and create a three-
dimensional form that determined the interaction
with the ligand. The docking simulation of all
processes involved in the preparation of the protein
structure was run in the YASARA program.
The process of preparing the ligand structure
aimed to achieve optimal ligand conformation. The
conformation of drug molecules may depend on the
acidity (pH) and ionic composition of the medium in
which the drug is studied (Siswandono and
Soekardjo 1998). Since the drug worked on a
biological system, each ligand was subjected to
protonation to obtain a structure adjusted to the
blood pH, i.e., about 7.4. A total of nine ligands
were tested in this study. The nine ligands consisted
of one endogenous ligand (SIM) from the crystal
structure of HMG-CoA reductase (for redocking
process), one comparator ligand (simvastatin), and
seven test ligands of the compounds in papaya seeds,
namely carpaine, benzyl isothiocyanate, benzyl
glucosinolate, glucotropaeolin, benzyl thiourea, and
oleic acid. Using a conformational search in Marvin
Sketch, the optimization yielded as many as ten (10)
conformations that represented the positions of all
ligands against the pocket cavity. Then, the analysis
proceeded with the docking process to find out
which ligand conformation best represented the
ligand position against the pocket cavity, as
evidenced by the ChemPLP score analysis. Agistia
et al. (2013) state that the most appropriate
conformation can be identified from the output of
the molecular docking run in PLANTS 1.2, which is
the ChemPLP score. Therefore, the next step in this
research determined the ChemPLP score of the
ligand against the receptor resulted from the docking
process to find out the best ligand conformation.
The identification of appropriate docking
protocols is a key step to a valid docking pose
(Oniga et al., 2017). The validation of the docking
method in this study was conducted by redocking
the endogenous ligand in the protein group
downloaded from the Protein Data Bank. The
evaluation of the validation results relied on the
RMSD (Root Mean Square Deviation) of the pose
visualization (Moitessier et al., 2008). RMSD is a
measurement of two poses by comparing the
positions of atoms in experimental structures with
the ones in docked or predicted structures (Hawkins
et al., 2008) The RMSD values of successful
docking methods are <2.0 Å (Hevener et al., 2009;
Jain and Nicholls, 2008; Moitessier et al., 2008).
The RMSD of the validation results in this research
was 1.0138 Å (<2 Å), proving that 1HW9 could be
used for further analysis in this research. The closer
the RMSD to zero, the more similar the poses of
endogenous ligand and copy ligand. Small RMSD
suggests that the developed protocols are accepted,
and they can be further developed for virtual
screening in the discovery of new compounds
(Purnomo, 2011; 2013). Superposing the
endogenous ligand and the copy ligand, the
visualization validated that the atoms of the two-
molecule structures had similar positions and angles
(Figure 2). In other words, the conformation of the
endogenous ligand structure of GDP was similar to
the well-selected copy ligand in the docking process
(Adelina, 2014). With RMSD >2.0 Å, the
visualization shows two molecules with significantly
different angles and positions even though they have
equal number of atoms.
Docking is a simulation method to find out the
orientation between ligand and receptor. After the
redocking process with endogenous ligand, the
cartesian coordinates of the binding site were x=
4.0308, y= -9.4318, and z= -11.5016. Figure 3
shows the visualization of the binding site on the
receptor (1HW9). The binding site is an area where
protein binds to molecules and ions (i.e., ligands)
that will affect the conformation and function of the
Figure 2. The Visualization of the Superposition of
Endogenous Ligand and Copy Ligand in Molegro
Molecular Viewer Software. The endogenous ligand is
yellow; the copy ligand is blue.
MICH-PhD 2018 - 1st Muhammadiyah International Conference on Health and Pharmaceutical Development
126

protein. The binding site involves amino acid
residues that play an important role in binding with
ligands (Pratama et al., 2016). Based on the docking
scores listed in Table 1, the endogenous SIM
(simvastatin acid) ligand has the lowest ChemPLP
score, i.e., -101.899 kcal/mol, while the score of
simvastatin (comparator ligand) is -80.3996
kcal/mol. Simvastatin acid is the active metabolite of
simvastatin (Pubchem, 2017). The difference lies in
the structure. Mycek et al. (2001) mention that the
structure of simvastatin is a lactone that needs to be
hydrolyzed into active drugs. This hydrolysis
(simvastatin acid) adds OH-group and carboxylic
group to the structure. The OH-group influences the
amount of hydrogen bond interaction. Siswandono
and Soekardjo (1998) explain that hydrogen bond
interaction generally occurs in compounds that have
clusters of, for example, OH-, and NH-. Based on
the data (Table 2), the most prevalent hydrogen bond
interactions in the endogenous ligand (simvastatin
acid) involved the OH group, especially OH- in the
carboxylic group of simvastatin acid. The difference
in the number of OH-groups causes different scoring
results between simvastatin and simvastatin acid.
The molecular docking performed on the
compounds of papaya seed against HMG-CoA
reductase (Table 1) resulted in three (3) best
compounds whose ChemPLP scores were lower than
the comparator ligands (simvastatin). They were
benzyl glucosinolate (ChemPLP score= -90.5491
kcal/mol), glukotropeolin (-85.1919 kcal/mol), and
oleic acid (-81.7665 kcal/mol). The ChemPLP score
of simvastatin ligand was -80.3996 kcal/mol.
Schneider and Bohm (2000) mention that a smaller
docking score implies a more stable bond or, in
other words, a more potent compound. Serina (2013)
affirms this assertion with the energy linkage to
affinity, i.e., that the best ligand will have stable
(free energy) performance and a better affinity.
Affinity is a measure of the drug's ability to bind
receptors. It is highly dependent on the molecular
structure of the drug and the receptor (Siswandono
and Soekardjo 1998). The ChemPLP scores of the
ligands in Table 1 were compared with simvastatin
to determine which ligand had the best interaction
and affinity. The comparison results were validated
by further analysis based on the inter-molecular
bond interactions (ligand and receptor). The analysis
also included the interpretation of the bond
interaction between amino acid residues and ligands
in Molegro Molecular Viewer (MMV).
Glucosinolate is included in the glycoside class.
A glycoside is composed of two entities, namely the
sugar group (glycone) and the non-sugar group
(aglycone/genin). The sugar portion of a glycoside
may be associated with the aglycone in various
ways, and the most common one is through the
oxygen (O-glycosides) atoms. However, the atoms
Figure 3. (A) Zoom Out, (B) Zoom in; The Visualization
of the Binding Site on 1HW9 (Grid Box: Green Circle)
using MMV Software Based on the Redocking Results in
PLANTS 1.2 software
.
(
A
)
(
B
)
Table 1. The Molecular Docking Results of the
Comparator Ligands and Ligands in Papaya Seed against
HMG-CoA Reductase Using PLANTS 1.2 Software
Ligands
CHEMPLP
Scores
(Kcal/mol)
Simvastatin Acid
(endogenous ligand) -101.899
Simvastatin
(Comparative Ligand) -80.3770
Carpaine
-66.6044
β-Sitosterol
-73.2094
Benzylthiourea
-55.9158
Benzyl Isothiocyanate
-52.0111
Benzyl Glucosinolate
-90.5491
Oleic acid
-81.7665
Glucotropaeolin
-85.1919
In Silico Analysis of the Phytochemical Compounds in Carica papaya Seeds for Optimizing the Inhibitors of HMG-CoA Reductase
127

that connect them may be Carbon (C-glycosides),
Table 2. The visualization Results of the hydrogen bond interaction between the ligands (endogenous ligands, test
ligands, and comparator ligands) and the receptor (HMG-CoA reductase) using MMV software
Ligands
Bonded
Amino
Acid
Residues
Bond
distance
(Å)
Group on
Ligands
Ligands
Bonded
Amino
Acid
Residues
Bond
distance
(Å)
Group on
Ligands
Simvastatin
Acid
Asp 767 2.75 OH- group
Glucotropaeolin Asp 767 2.97 Nitro
g
rou
p
Gln 770 3.30 OH-group
on
carboxylate
Gln 766 3.18
Sulfonic
group
Gln 770 3.18 OH-group
on
carboxylate
Ser 774 2.40 OH
-
group on
glucose
Glu 801 3.12 O- pada
carboxylate
Ser 774 3.24 OH
-
group on
g
lucose
Asp 690 3.14 OH-group
on
carbox
y
late
Gln 770 2.76 OH
-
group on
g
lucose
Asp 690 3.37 OH-group
on
carbox
y
late
Gln 770 2.60 OH
-
group on
g
lucose
Asn 771 3.03 Side chains
Gln 770 3.11 OH
-
group on
glucose
Arg 702 3.26 OH-group
on
carboxylate
Tyr 761 2.79 OH
-
group on
glucose
Simvastatin Tyr 761 2.85 Lactone
ring
Glu 801 2.62 OH
-
group on
g
lucose
Gln 770 2.77 Side chains
Glu 801 3.24 OH
-
group on
g
lucose
Gln 766 3.26 OH- group
on
lactone’s
rin
g
Asn 771 3.37 OH
-
group on
glucose
Benzyl
glucosinolate
Lys 691 2.15 OH
-
group
on glucose
Ala 769 3.27 O
-
on
glucose
rin
g
Asp 767 2.66 OH
-
group
on glucose
Oleic acid Arg 702 3.14 OH-
group
Gln 770 2.51 OH
-
group
on glucose
Asp 690 3.11 OH-
group
Gln 770 2.41 OH
-
group
on
g
lucose
Asp 690 2.89 OH-
g
rou
p
Glu 801 2.39 OH
-
group
on
g
lucose
Glu 801 2.46 OH-
g
rou
p
Ser 774 2.95 Nitro
group on
g
lucose
Glu 801 2.82 OH-
group
Tyr 761 2.32 Sulfonic
group
Tyr 761 2.77 Sulfonic
group
MICH-PhD 2018 - 1st Muhammadiyah International Conference on Health and Pharmaceutical Development
128
Nitrogen (N-glycosides), or sulfur atoms (S-
glycosides) (Sarker and Nahar, 2009). In the
glucosinolate compound, the sugar portion (glycone)
is connected to a sulfur atom (S-glycoside).
Glucosinolate is a secondary metabolite of almost all
Brassicales families (including Brassicaceae,
Capparidaceae, and Caricaceae) (James et al., 1996).
One example of glucosinolate found in the
Caricaceae family discussed in this study is
glucosinolate, derived from the benzyl glucosinolate
(
Figure 4) in papaya seed. Investigating the benzyl
glucosinolate content in various tissues, Najamura et
al. (2007) find the highest benzyl glucosinolate
content in papaya seeds.
The receptor’s interaction with ligands formed
after the docking process was visualized using
Molegro Molecular Viewer software. The breaking
lines described the hydrogen bonds that occurred
between the residues and the groups on the ligands.
The observation of residual interactions (amino
acids) aimed to identify any ligand-receptor
interactions. The hydrogen bonding is an interaction
that can stabilize the ligand bond and the receptor
bond. Another ligand-receptor interaction that can
improve the stability of the conformation is the
electrostatic interaction and van der Walls
interactions.
Table 2 shows the residual ratio of the two best
ligands to simvastatin (comparator ligand) after the
docking process. Simvastatin bonded to Tir 761, Gli
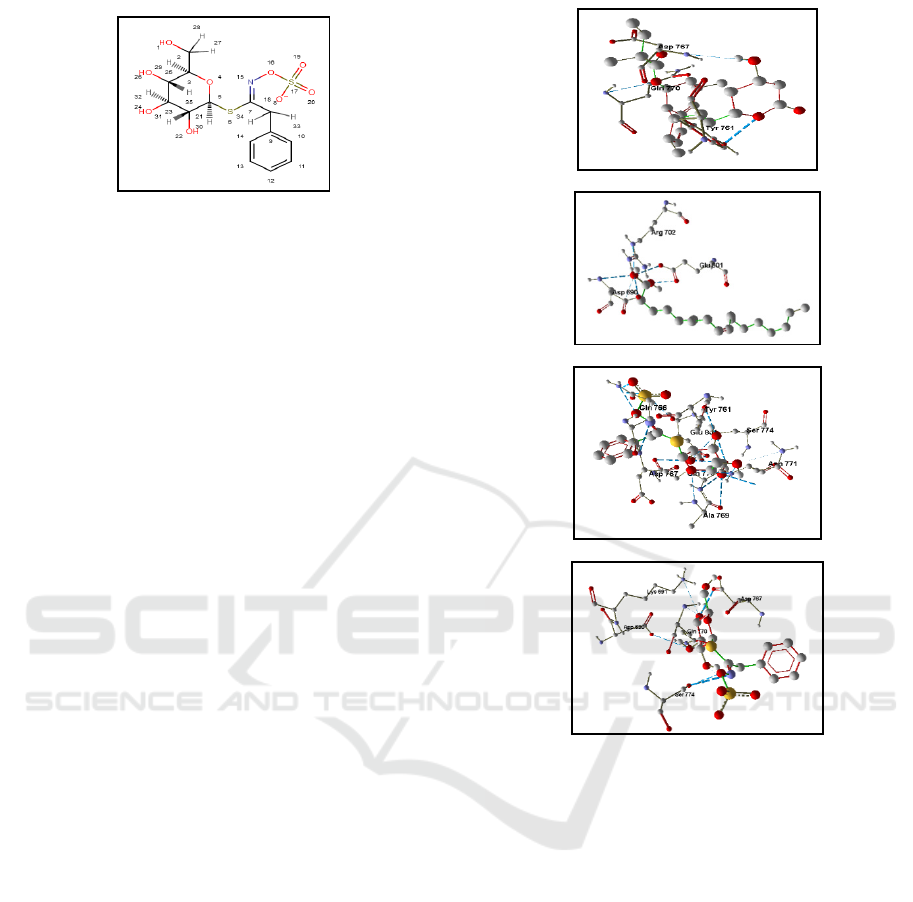
770, and Gli 766 (Figure 5a). The number of the
hydrogen bonds formed in simvastatin tended to be
lower than those of oleic acid, glucotropaeolin, and
benzyl glucosinolate. The hydrogen bond distances
formed in this research were greater than 3.0 Å
(close to 3.5 Å), but not one of them exceeded 3.5 Å.
Therefore, the hydrogen bond distance of
simvastatin still qualifies for an energetically
significant hydrogen bond interaction, i.e., not
exceeding 3.5 Å (Marcou and Rognan, 2007). In the
interaction between oleic acid ligands and HMG-
CoA reductase receptors, there were five hydrogen
bond interactions (Figure 5d and Table 2). The
hydrogen bonds occurred in the amino acid residue
Arg 702 (1 hydrogen bond interaction), Asp 690 (2
hydrogen bond interactions), and Glu 801 (2
hydrogen bond interactions). Two of the five
hydrogen bonds in oleic acid had the hydrogen bond
distance of greater than 3.0 Å (<3.5 Å), which meets
the requirement of significant hydrogen bond
(Marcou and Rognan, 2007). When compared with
simvastatin as a comparator ligand in this research
(Table 2), oleic acid had a greater number of
hydrogen bond interactions. This finding is in line
with Natali et al. (2007) who report that oleic acid
exhibits an inhibition activity toward the enzyme
HMG-CoA reductase.
Figure 4. The Structure of Benzyl Glucosinolate
(Chemicalize, 2018)
(A)
(B)
(C)
(D)
Figure 5: The Visualization of The Residual Contact
of Ligand and HMG-CoA Reductase Receptor using
Molegro Molecular Viewer Software: (A) Simvastatin,
(B) Oleic acid, (C) Glucotropaeolin, (D) Benzyl
glucosinolate.
In Silico Analysis of the Phytochemical Compounds in Carica papaya Seeds for Optimizing the Inhibitors of HMG-CoA Reductase
129

There were twelve hydrogen bond interactions in
the glucotropaeolin ligand (Figure 5c and Table 2).
Most hydrogen bond interactions were formed due
to a large number of electronegative atoms in the
glucotropaeolin molecule; hence, the tendency to
form hydrogen bonds. However, according to
Lipinski (2003), the number of hydrogen bonds in
the drug should not be more than ten. Otherwise,
drugs will have difficulty in passing through the
intestinal walls into the blood.
Pine et al. (1988) mention that the shorter the
bond distance, the stronger the bond. The
interactions in glucotropaeolin included six (6)
hydrogen bonds whose distances were more than 3.0
Å, i.e., in Ala 769 (3.27 Å), Asn 771 (3.37 Å), Glu
801 (3.24 Å), Gln 770 (3.11 Å), Ser 774 (3.24 Å),
and Gln 766 (3.18 Å). According to Marcou and
Rognan (2007), hydrogen interactions can occur
when two atoms are within 3.5 Å to each other. The
hydrogen bond distance in glucotropaeolin was
smaller than 3.5 Å, which satisfies the conditions for
hydrogen bonding.
Glucotropaeolin was one of the three (3) best
ligands in papaya seeds with a lower ChemPLP
score than simvastatin (Table 1). However, after
further analysis through visualization, each of its
hydrogen bond distances satisfied the conditions
hydrogen bonding (i.e., <3.5 Å) (Marcou and
Rognan, 2007). However, it is thought to be less
potent in penetrating the intestinal membrane
because it does not meet the requirement proposed
by Lipinski (2003), i.e., the number of hydrogen
bonds should not be more than ten. Considering the
number of hydrogen bond interactions,
glucotropaeolin ligand could not be categorized as
the best ligand.
Once proven by visualization, the number of
hydrogen bond interaction between benzyl
glucosinolate and amino acid residues at the receptor
(Figure 6) was eight. This number was greater than
the hydrogen bond interaction in simvastatin.
Additionally, it conforms with the qualification set
in Lipinski (2003), i.e., not exceeding 10. The amino
acid residues that interacted with benzyl
glucosinolate were Lys 691 (with a hydrogen bond
distance of 2.15 Å), Asp 767 (2.66), Gln 770 (2.51 Å
and 2.41 Å), Glu 801 (2.39 Å), Ser 774 (2.95 Å),
and Tyr 761 (2.32 Å and 2.77 Å). The average
length of the hydrogen bond on benzyl glucosinolate
ligand was less than 3.0 Å, which is in line with the
conditions for hydrogen bonding mentioned in
Marcou and Rognan (2007). The average length of
the hydrogen bond distance formed on benzyl
glucosinolate was shorter than the comparator
ligands (simvastatin) and the other two best ligands
in papaya seeds (glucotropaeolin and oleic acid)
(Table 2). The shorter the hydrogen bond distance,
the stronger the bond (Pine et al., 1988).
Drug-likeness is a qualitative concept used to
describe the similarity of a compound as a drug
candidate, such as the complex balance of various
molecular properties and structural features that
determine whether a particular molecule is similar to
a known drug. These molecular properties are
primarily hydrophobicity, electronic distribution,
hydrogen bond characteristics, molecular size and
flexibility, and other pharmacophore properties
affecting the behavior of molecules in living
organisms, including bioavailability, delivery
properties, affinity for proteins, reactivity, toxicity,
and other metabolic stability (Leeson, 2016; Mishra
et al., 2017). The Rule of Five (Ro5) or the
Lipinski’s Rule of Five is a set of in silico guidelines
applied to drug discovery to prioritize compounds
with a high probability of increased absorption
(Doak et al., 2014). This rule can be used to
determine the pharmacokinetics of a compound as a
drug candidate (Benet et al., 2016). For drug-
likeliness evaluation, it discusses four simple
physicochemical parameters (namely, molecular
weight ≤ 500, log P ≤ 5, hydrogen bond donor ≤ 5,
hydrogen bond acceptor ≤ 10) associated with 90%
of orally active drugs that have passed clinical status
of phase II (Lipinski, 2004; 2016).
Based on the prediction results (Table 3) run in
www.chemicalize.com using the Lipinski’s Rule of
Five, benzyl glucosinolate was within the threshold
of the partition coefficient (log P= 2.19; <5). The log
P values of benzyl glucosinolate and glucotropaeolin
were lower than the endogenous compounds and
ligands, but they still met the Lipinski’s rule (log P
<5). The log P values of benzyl glucosinolate and
glucotropaeolin indicated a solubility coefficient in
Table 3. The Prediction Results Based on the Lipinski’s
Rule of Five (Chemicalize, 2018)
Ligands
Prediction using the Lipinski’s
Rule of Five
BM
(g/mol
)
Log
P
H
bond
Dono
r
H
bond
A
cce
p
to
r
Simvastatin 418.57 4.46 1 3
Benzyl
Glucosinolate
408.42 2.19 4 9
Oleic aci
d
282.47 6.78 1 2
Glucotro
p
aeolin 409.42 2.19 5 9
Simvastatin aci
d
436.59 3.9 3 6
MICH-PhD 2018 - 1st Muhammadiyah International Conference on Health and Pharmaceutical Development
130

fat or water within the range of -0.4 and 5. The
molecular weights of benzyl glucosinolate, oleic
acid, and glucotropaeolin were 408.42 g/mol, 282.47
g/mol, and 409.42 g/mol (<500 g/mol), and they
were lower than the BM of the comparator ligand.
The hydrogen bond donors in benzyl
glucosinolate and oleic acid were, respectively, 4
and 1 (<5). Meanwhile, glucotropaeolin had 5
hydrogen bond donors, which did not meet the
Lipinski’s rule. The number of the hydrogen bond
donors in benzyl glucosinolate was higher than the
comparator ligand, but it still met the standards set
by Lipinski (1997). Benzyl glucosinolate and
glucotropaeolin had 9 hydrogen bond acceptors
(close to 10), while oleic acid had two (2). However,
the Lipinski’s rule states that the hydrogen bond
acceptor should not exceed 10. As a conclusion, the
hydrogen bond donors and acceptors in benzyl
glucosinolate, oleic acid, and glucotropaeolin are
compliant with the Lipinski’s rule.
This rule also states that a molecular weight of
more than 500 Da cannot diffuse through the cell
membrane by passive diffusion. The higher the log
P, the more hydrophobic the molecule. Molecules
that have too hydrophobic properties tend to have
high levels of toxicity because they will stay longer
in the lipid bilayer and spread more widely in the
body; therefore, the selectivity of the bond to the
target enzyme decreases. Too hydrophilic properties
(negative log P) are also not good because the
molecule cannot pass through the membrane lipid
bilayer. Based on the number of hydrogen bond
donor and acceptor, a higher hydrogen bonding
capacity represents larger energy required for the
absorption process to occur. In general, the
Lipinski’s rules describe the solubility of certain
compounds to penetrate cell membranes by passive
diffusion (Lipinski et al., 1997). As a conclusion,
benzyl glucosinolate does not violate any of the
Lipinski's rules, and, thereby, it can be developed as
anti-hypercholesterolemia drug candidates for oral
preparations.
4 CONCLUSIONS
The results of the ligand-receptor interaction of the
compounds in papaya seed (Carica papaya L.)
against HMG-CoA reductase receptor categorized
benzyl glucosinolate as the best compound because
it had a ChemPLP score of -90.5491 kcal/mol and
eight hydrogen bond interactions. This compound
has the potential as an anti-hypercholesterolemia
drug.
ACKNOWLEDGMENTS
Authors would like to gratefully appreciate the
Indonesian Ministry of Research, Technology, and
Higher Education for their support during the
research.
REFERENCES
Adelina R. 2014. Uji Molecular Docking Annomuricin E
dan Muricapentocin pada Aktivitas Antiproliferasi
(Molecular Docking Studies of Annomuricin E and
Muricapentocin on Antiproliferation Activity ). Jurnal
Ilmu Kefarmasian Indonesia. 12(1): 32–36.
Afrose S, Hossain S, Salma U, Miah AG, Tsujii H. 2010.
Dietary Karaya Saponin and Rhodobacter capsulatus
Exert Hypercolestolemics Effect by Suppression of
Hepatic Cholesterol Synthesis and Promotion of Bile
Acid Synthesis in Laying Hens. Research Article. p. 1-
7.
Agistia DD, Purnomo H, Tegar M, Nugroho AE. 2013.
Interaksi Senyawa aktif dari (Aegle marmelos) Correa.
Sebagai Anti Inflamasi dengan Reseptor Cox-1 dan
Cox2. Journal of Traditional Medicine. 18(2):80-87.
Benet L Z, Hosey CM, Ursu O, Oprea TI. 2016. BDDCS,
the Rule of 5 and drugability. Advanced Drug Delivery
Reviews. 101: 89–98.
Chemicalize. 2018. Calcutaion Structure of Simvastatin,
Benzyl Glucosinolate, oleic acid, Glucotropaeolin and
Simvastatin acid. www.chemicalize.com. Diakses 2
Agustus 2018.
Doak B, Over B, Giordanetto F, Kihlberg J. 2014. Oral
Druggable Space beyond the Rule of 5: Insights from
Drugs and Clinical Candidates. Chemistry & Biology.
21(9): 1115–1142.
Goldstein, J.L., Brown, M.S. 1990 Regulation of the
mevalonate pathway. Nature, 343, 425-430.
Hawkins P, Warren G, Skillman A, Nicholls A. 2008.
How to do an Evaluation: Pitfalls and Traps. Journal
of Computer-Aidid Molecular Design. 22(3): 179–190.
Jain A, Nicholls A. 2008. Recommendations for
Evaluation of Computational Methods. Journal of
Computer-Aidid Molecular Design. 22: 133–139.
James ER, Kenneth GK, Robert AP, Kenneth JS. 1996.
Molecules Morphology and Dahlgren’s Expanded
Order Capparales. Jurnal Systematic Botany.
21(3):289.
Kitchen DB, Decornez H, Furr JR, Bajorah J. 2004.
Docking and Scoring in Virtual Screening for Drug
Discovery: Methods and Applications. Journal of
Nature Reviews Drug Discovery. 3(11): 935-949.
Leeson P. 2016. Molecular Inflation, Attrition and the
Rule of Five. Advanced Drug Delivery Reviews. 101:
22–23.
Lipinski CA. 2003. Physicochemical Properties and The
Discovery Of Orally Active Drugs: Technical And
In Silico Analysis of the Phytochemical Compounds in Carica papaya Seeds for Optimizing the Inhibitors of HMG-CoA Reductase
131

People Issues. Journal of Molecular Informatics:
Confronting Complexity. Hlm. 1-20.
Lipinski C. 2004. Lead-and Drug-like Compounds: The
Rule-of-Five Revolution. Drug Discovery Today:
Technologies. 1(4): 337–341.
Lipinski C. 2016. Rule of Five in 2015 and Beyond:
Target and Ligand Structural Limitations, Ligand
Chemistry Structure and Drug Discovery Project
Decisions. Advanced Drug Delivery Reviews. 101: 34–
41.
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. 1997.
Experimental and Computational Approaches to
Estimate Solubility and permeability in drug discovery
And Development Settings. Journal of Advanced Drug
Delivery Reviews. 23(1-3):3-25.
Marcou G, Rognan D. 2007. Optimizing Fragment and
Scaffold Docking by Use of Molecular Interaction
Fingerprints. Journal of Chemical Information and
Modelling. 47(1):195-207.
Mishra SS, Sharma CS, Singh HP, Pandiya H. 2017. In
Silico Pharmacokinetic and Toxicity Study of Some
Selected Antidepressant Drugs. Chemistry Research
Journal. 2(1): 42–45.
Moitessier N, Englebienne P, Lee D, Lawandi J, Corbeil
C. 2008. Towards the Development of Universal, Fast
and Highly Accurate Docking Scoring Methods: a
Long Way to Go. British Journal of Pharmacology.
153(1): 7–26.
Mycek MJ, Harvey RA, Champe PC. 2001. Farmakologi
Ulasan Bergambar Edisi II Cetakan I. Widya Medika.
Jakarta. Hlm 216.
Nakamura Y, Yoshimoto M, Murata Y, Shimoishi Y, Asai
Y, Park EY. 2007. Papaya Seed Represents A Rich
Source of Biologically Active Isothiocyanate. Journal
of Agricultural and Food Chemistry 55(11): 4407-
4413.
Nakanishi M, Goldstein JL, Brown MS. 1988. Multivalent
Control of 3-hidroxy-3-methylglutaryl Coenzyime A
Reductase. Mevalonate Derived Product Inhibits
Translations of mRNA and Accelerates Degradation
of Enzyme. Journal of Biological Chemistry. 263(18):
8929-37.
Natali F, Siculella L, Salvati S, Gnoni GV. 2007. Oleic
Acid is A Potent Inhibitor of Fatty Acid and
Cholesterol Synthesis in C6 Gliomca Cells. Journal of
Lipid Resolution. 48(9):1966-1975.
Olivera T, Ricardi KFS, Almeida MR, Costa MR, Nagem
TJ. 2007. Hypolipidemic Effect of Flavonoids and
Cholesterolamine in Rats Tania. Latin American
Journal of Pharmacy. 26(3): 407-410.
Oniga SD, Pacureanu L, Stoica CI, Palage MD, Crăciun
A, Rusu LR, Araniciu C. 2017. COX inhibition profile
and molecular docking studies of some 2-
(Trimethoxyphenyl)-Thiazoles. Molecules. 22(9): 1–
15.
Pine SH, Hendrickson JB, Cram DJ, Hammond GS. 1988.
Kimia Organik 2 Jilid I, II. Terjemah: Roehyati J,
Sasanti WP. Institut Teknologi Bandung. Bandung. p.
93, 899.
Pratama MFY, Abidi SR, Firdaus KA, Aulia AF, Santoso
B. 2016. Kajian Docking Molekular Pada Binding Site
Pocket Dari Plavopiridol Dalam Menghambat
Glikogenfosforilase Menggunakan Pyrx-Autodock
Vina. Jurnal of The Fourth University Research
Coloquium. Hlm. 154-158.
PubChem. 2017. Benzyl Glucosinolate, Benzyl Thiourea.
https://www.ncbi.nlm.nih.gov. Accessed on August
19, 2017.
Purnomo H. 2011. Kimia Komputasi Molecular Docking
PLANTS. Yogyakarta: Pustaka Pelajar.
Purnomo H. 2013. Kimia Komputasi untuk Farmasi dan
Ilmu Terkait. Pustaka Pelajar. Yogyakarta. p. 17-80.
Sarker SD, Nahar L. 2009. Kimia Untuk Mahasiswa
Farmasi. Yogyakarta: Pustaka Pelajar. p. 15, 39, 41,
42, 44.
Satriyasa BK, Pangkahila W, 2010. Fraksi Heksan dan
Fraksi Metanol Ekstrak Biji Pepaya Muda
Menghambat Spermatogonia Mencit (Mus Musculus)
Jantan. Jurnal Veteriner. Denpasar-Bali. 11(1): 37-39.
Schneider G, Bohm J. 2000. Virtual Screening for
Bioactive Molecules. WILEY-VHC. Weinheim: 1-
308.
Serina JJC. 2013. Enzymatic Inhibitory Activity of
Hydroxycinnamates (HCs) In Silico Studies.
Universidade de madeira. Portugal. Master
Thesis.:112.
Siregar. 2015. The Effect Eugenia polyantha Extract on
LDL Cholesterol. Review. Universitas Lampung. 4(5):
85-92.
Siswandono, Soekardjo. 1998. Prinsip-prinsip Rancangan
Obat. Surabaya: Airlangga University Press. p. 65, 82,
91, 94.
Stapleton PA, Goodwill AG, James ME, Brock RW,
Frisbee JC. 2010. Hypercholesterolemia and
microvascular dysfunction: interventional strategies.
Journal of Inflammation. 7:54
Yogiraj V, Pradeep KG, Chetan SC, Anju G, Bhupendra
V. 2014. Carica papaya L: An Overview.
International Journal of Herbal Medicine. 2(5): 01-08.
Zukrullah M, Aswad M, Subehan. 2012. Kajian Beberapa
Senyawa Antiinflamasi Docking Terhadap
Siklooksigenase-2 Secara in Silico
. Majalah Farmasi
dan Farmakologi. 16(1): 37-44.
MICH-PhD 2018 - 1st Muhammadiyah International Conference on Health and Pharmaceutical Development
132
