
Towards the Prokaryotic Regulation Ontology: An Ontological Model to
Infer Gene Regulation Physiology from Mechanisms in Bacteria
Citlalli Mej
´
ıa-Almonte
a
and Julio Collado-Vides
b
Center for Genomic Science, UNAM, Av. Universidad, Cuernavaca, Mexico
Keywords:
Formal Ontology, Domain Ontology, Gene Regulation, Bacteria.
Abstract:
Here we present a formal ontological model that explicitly represents regulatory interactions among the main
objects involved in transcriptional regulation in bacteria. These formal relations allow the inference of gene
regulation physiology from gene regulation mechanisms. The automatically instantiated classes can be used
to assist in the mechanistic interpretation of gene expression experiments done at the physiological level, such
as RNA-seq. This is the first step to develop a more comprehensive ontology focused on prokaryotic gene
regulation. The ontology is available at https://github.com/prokaryotic-regulation-ontology
1 INTRODUCTION
Since the success shown by the Gene Ontology as
a controlled vocabulary, bio-ontologies are increas-
ingly important tools in bio-informatics. However,
little has been explored regarding formal ontological
representation in the domain of bacterial gene reg-
ulation. There are two granularity levels at which
gene regulation can be studied. At the physiological
level, transcript concentration or gene product activity
is directly measured under some condition, normally
adding or depleting certain chemicals to growth me-
dia (Burstein et al., 1965). At the mechanistic level,
the effect of specific mutations on gene expression is
studied to discover the precise regulators involved in
some system (Ptashne, 1967). At this level, the most
studied mechanisms are those of transcription initia-
tion mediated by transcription factors. These proteins
can adjust gene expression to environmental require-
ments using their two main functional domains: the
effector-binding domain that senses the environmen-
tal signal and the DNA binding domain. Transcription
factors bind to DNA in sites called transcription factor
binding sites, thereby increasing or decreasing the ac-
tivity of a promoter. Promoters are the DNA regions
where transcription of transcription units (TUs) be-
gins; TUs in turn contain one or more genes. There-
fore, the expression of a TU is regulated by regula-
tion of the promoter activity. Here, we develop an
ontological model that can infer the physiology from
a
https://orcid.org/0000-0002-0142-5591
b
https://orcid.org/0000-0001-8780-7664
mechanisms of gene regulation.
The result of transcriptome analysis are sets of
genes that are either underexpressed or overexpressed
under a given condition, including the addition of
chemicals to growth media. The observation of un-
derexpression corresponds to the observation of gene
inhibition, whereas the observation of overexpres-
sion corresponds to the observation of gene induc-
tion. This means that transcriptome analysis gives us
physiological insights, rather than mechanistic ones.
The model presented here, automatically instantiates
sets of genes that are induced or repressed by some
molecule based on the mechanisms of induction or re-
pression. The final terms will encode both the physi-
ology and the mechanisms of gene regulation (see be-
low). Thus, this ontology can help in the mechanistic
interpretation of gene expression experiments that are
done at the physiological level, such as transcriptome
analysis.
No ontology explicitly states the aim of mod-
eling gene regulation in the obo-foundry reposi-
tory (Smith et al., 2007); whereas a search in bio-
portal (Noy et al., 2009; Noy et al., 2001) only re-
trieves the Gene Regulation Ontology (GRO) (Beis-
swanger et al., 2008). This ontology includes object
properties to define agents and patients of regulation,
but it focuses on the mechanistic description of gene
regulation and it does not distinguishes the two granu-
larity levels of gene regulation described in this paper.
Thus, here we develop an ontology to represent both
mechanisms and physiology of gene regulation, the
later inferred from the former.
Mejía-Almonte, C. and Collado-Vides, J.
Towards the Prokaryotic Regulation Ontology: An Ontological Model to Infer Gene Regulation Physiology from Mechanisms in Bacteria.
DOI: 10.5220/0008387804950499
In Proceedings of the 11th International Joint Conference on Knowledge Discovery, Knowledge Engineering and Knowledge Management (IC3K 2019), pages 495-499
ISBN: 978-989-758-382-7
Copyright
c
2019 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
495

2 DEVELOPMENT PROCESS
We are using top-down ontology development ap-
proach (Noy et al., 2001). First, we included the most
general and important entities involved in regulation
of transcription initiation: transcription factor, tran-
scription factor binding site, promoter, transcription
unit, effector, etc. Second, we included the corre-
sponding biological relations among them. Third, we
created the classes that will be automatically instan-
tiated: TF bound to the DNA and regulated system.
Fourth, we formally defined these classes taking ad-
vantage of the biologically relations included in the
second step (figure 2). Lastly, most specific terms
have to be generated for each specific TF, TU, pro-
moter, etc. along with their relations. The model will
automatically classify these specific entities into the
defined classes (see glycolate example). RegulonDB
can be used to instantiate the ontology with knowl-
edge about Escherichia coli K-12 (Santos-Zavaleta
et al., 2018).
We are following the OBO-foundry princi-
ples. For this, we are taking advantage of the
OBO tools ROBOT (Overton et al., 2015) and
the Ontology Development Kit (https://github.com/
INCATools/ontology-development-kit). The first one
is mainly used to extract terms and modules from ex-
isting ontologies, while the later is designed for stan-
dardized ontology documentation and release of OBO
ontologies, taking care of quality control issues. We
are using the Basic Formal Ontology as upper-level
ontology. So far, we have reused terms from six OBO-
foundry ontologies: CHEBI, GO, MSO, NCIT, OGG,
and SO (Ashburner et al., 2000; de Matos et al., 2010;
Mungall et al., 2011; Sioutos et al., 2007; He et al.,
2014) The creation of new classes and axioms was
done using Prot
´
eg
´
e version 5.5. (Musen et al., 2015)
3 MODEL DESCRIPTION
In this paper, classes are written in italics and object
properties are written in bold face. Hierarchy is rep-
resented as indentation of bulleted lists.
3.1 An n-ary Relation to Represent the
Central Transcriptional Regulatory
Interaction
Figure 1 depicts the main elements involved in tran-
scriptional regulation along with the relations that
exist among them. These were ontologically repre-
sented as follows. Transcription factor (TF), TF bind-
ing site (TFBS), effector, and functional conformation
classes were created. Then, an n-ary relation design
pattern was used to link these four elements (Noy and
Rector, 2004). TF bound to TFBS class was cre-
ated with four properties: has binding transcription
factor, has target TFBS, is realized in functional
conformation, and has effector (Figure 1).
3.2 A Property Chain to Infer
Regulation from Anatomy
Figure 1 also depicts how the two key relations that
distinguish physiology from mechanisms of transcrip-
tional regulation were ontologically represented. The
mechanistic level describes the direct effect that a TF
bound to a TFBS has over its cognate promoter, while
the physiological level describes the effect that the
environmental condition (in our current model repre-
sented by the effector molecule) has over the expres-
sion of genes in a transcription unit. Promoter and
transcription unit classes were created. Then tran-
scription unit was related with promoter using the
property is transcribed from, whereas promoter was
related to the class TF bound to TFBS with the prop-
erty has activity regulated by. The has expression
regulated by property was created along with the fol-
lowing rule chain expressed in functional syntax (Fig-
ure 1) (Hitzler et al., 2009):
SubObjectPropertyOf(
ObjectPropertyChain( :is transcribed from
:has activity regulated by )
:has expression regulated by
)
This rule chain represents the fact that if a TU is
transcribed from a promoter, and this promoter has its
activity regulated by a TF bound to a TFBS, then this
TF bound to a TFBS regulates the expression of the
TU.
3.3 Automatic Classification of
Regulated Systems
At the physiological level, there are only two possi-
bilities: induction or inhibition of gene expression.
At the mechanistic level, there are four possibilities.
Transcription factors bind to their cognate TFBSs and
regulate transcription only when they are in func-
tional conformation. Induction can be achieved by
activation when the binding of the effector activates
a transcription factor that increases the expression of
a TU (active conformation of TF is holo), or by de-
repression when the binding of the effector deacti-
vates a transcription factor that decreases the expres-
sion of a TU (active conformation of TF is apo). In-
hibition can be achieved by repression when the bind-
KEOD 2019 - 11th International Conference on Knowledge Engineering and Ontology Development
496
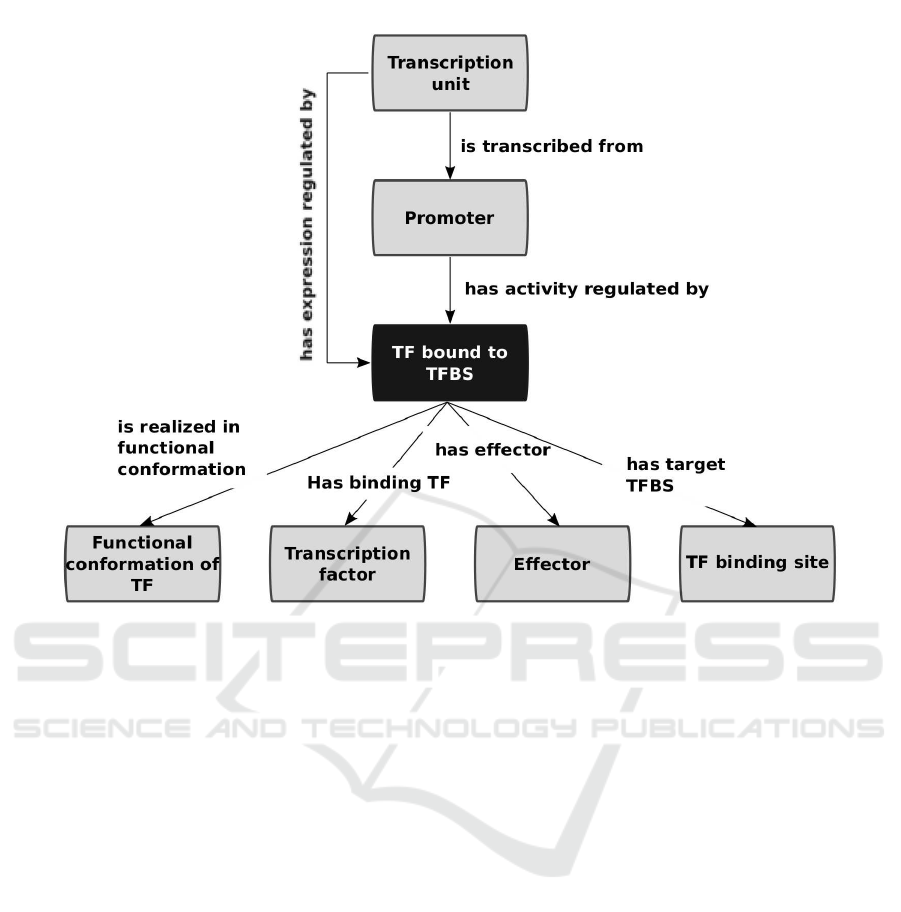
Figure 1: An n-ary relation and a property chain to represent the central regulatory interaction.
ing of the effector activates a transcription factor that
decreases the expression of a TU (active conforma-
tion is holo), or by de-activation when the binding of
the effector deactivates a transcription factor that in-
creases the expression of a TU (active conformation
is apo) (Balderas-Mart
´
ınez et al., 2013). All of these
cases describe the physiological response to the ap-
pearance of the effector. The disappearance of the ef-
fector reverses the response. We will treat these cases
later.
Therefore, to automatically classify TUs that are
induced or inhibited by an effector we have have cre-
ated the following subclasses of TF bound to TFBS
(Figure 2). Equivalent class axioms are shown.
• TF bound to TFBS in apo conformation is real-
ized in functional conformation some apo func-
tional conformation of TF
– TF-glycolate active in apo has effector some
glycolate
• TF bound to TFBS in holo conformation is real-
ized in functional conformation some holo func-
tional conformation of TF
– TF-glycolate active in holo has effector some
glycolate
The classes inducible system and inhibitable sys-
tem were created with the following subclasses.
Equivalent class axioms are shown.
• System induced by activation has expression in-
creased by some transcription factor bound to
TFBS in holo conformation
– System induced by activation by glycolate has
expression increased by some TF-glycolate
active in holo
• System induced by derepression has expression
decreased by some transcription factor bound to
TFBS in apo conformation
– System induced by derepression by glyco-
late has expression decreased by some TF-
glycolate active in apo
• System inhibited by repression has expression de-
creased by some transcription factor bound to
TFBS in holo conformation
– System inhibited by repression by glycolate has
expression decreased by some TF-glycolate
active in holo
Towards the Prokaryotic Regulation Ontology: An Ontological Model to Infer Gene Regulation Physiology from Mechanisms in Bacteria
497
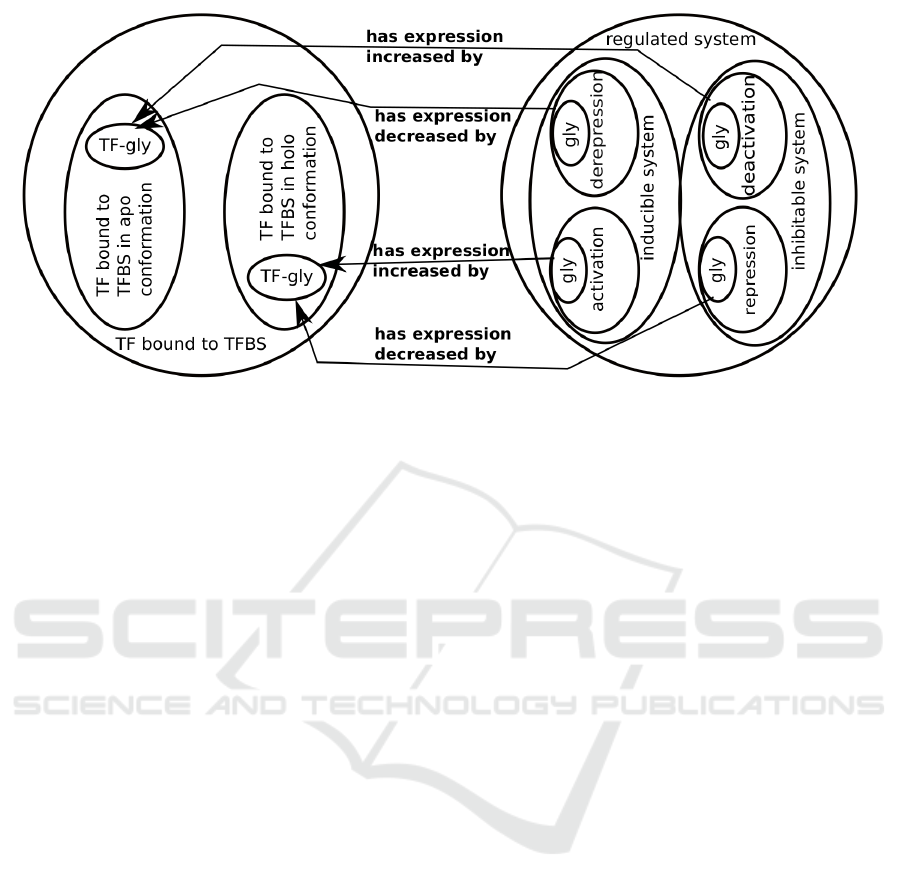
TF bound to TFBS
TF bound to
TFBS in holo
conformation
TF-gly
regulated system
TF bound to
TFBS in apo
conformation
TF-gly
inducible system
activation
gly
Figure 2: Defined classes to infer physiology from mechanisms. The outer circles represent the most general classes and
inner ovals more specific classes. On the left, the hierarchy of the molecular complex TF-TFBS-effector classes is shown.
In the text, the most specific classes are named TF-glycolate active in holo and TF-glycolate active in apo; in the figure, the
terms were shortened as TF-gly due to space issues. These classes are automatically instantiated due to the n-ary relation
shown in Figure 1. On the right, the hierarchy of effector-induced or effector-repressed systems are shown. The terms were
shortened due to space issues: activation is short for system induced by activation, derepression is short for system induced
by derepression, deactivation is short for system inhibited by deactivation, and repression is short for system inhibited by
repression, whereas gly is short for system induced by activation by glycolate, system induced by derepression by glycolate,
system inhibited by deactivation by glycolate, and system inhibited by repression by glycolate, depending on the superclass.
These classes can be automatically instantiated due to the property chain shown in Figure 1.
• System inhibited by deactivation has expression
increased by some transcription factor bound to
TFBS in apo conformation
– System inhibited by deactivation by glyco-
late has expression increased by some TF-
glycolate active in apo
In this listing of formal definitions, we included
only examples of classes defined by the specific effec-
tor glycolate. The final ontology have to be extended
to include classes for all known effectors. We plan to
do this extension using E. coli information retrieved
from RegulonDB.
4 CONCLUSIONS
An ontological model that can automatically classify
transcription units as effector-dependent repressible
or inducible systems was developed. This adds a layer
of formal knowledge to the mechanistic representa-
tion of bacterial gene regulation included in databases
like RegulonDB.
ACKNOWLEDGEMENTS
C.M.A. is a Ph.D. student from the Programa de Doc-
torado en Ciencias Biomedicas, Universidad Nacional
Autonoma de Mexico, receives fellowship 576333
from CONACYT and received financial aid from Pro-
grama de Apoyos para Estudios de Posgrado (PAEP)
for this conference. JCV acknowledges support by
UNAM and by NIH-NIGMS grant RO1-GM110597.
REFERENCES
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D.,
Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K.,
Dwight, S. S., Eppig, J. T., et al. (2000). Gene ontol-
ogy: tool for the unification of biology. Nature genet-
ics, 25(1):25.
Balderas-Mart
´
ınez, Y. I., Savageau, M., Salgado, H., P
´
erez-
Rueda, E., Morett, E., and Collado-Vides, J. (2013).
Transcription factors in escherichia coli prefer the
holo conformation. PloS one, 8(6):e65723.
Beisswanger, E., Lee, V., Kim, J.-J., Rebholz-Schuhmann,
D., Splendiani, A., Dameron, O., Schulz, S., Hahn, U.,
et al. (2008). Gene regulation ontology (gro): design
principles and use cases. In MIE, pages 9–14.
Burstein, C., Cohn, M., Kepes, A., and Monod, J. (1965).
Role du lactose et de ses produits metaboliques dans
KEOD 2019 - 11th International Conference on Knowledge Engineering and Ontology Development
498

l’induction de l’operon lactose chez escherichia coli.
Biochimica et Biophysica Acta (BBA)-Nucleic Acids
and Protein Synthesis, 95(4):634–639.
de Matos, P., Dekker, A., Ennis, M., Hastings, J., Haug,
K., Turner, S., and Steinbeck, C. (2010). Chebi: a
chemistry ontology and database. Journal of chemin-
formatics, 2(1):P6.
He, Y., Liu, Y., and Zhao, B. (2014). Ogg: a biological on-
tology for representing genes and genomes in specific
organisms. In ICBO, pages 13–20. Citeseer.
Hitzler, P., Kr
¨
otzsch, M., Parsia, B., Patel-Schneider, P. F.,
and Rudolph, S. (2009). Owl 2 web ontology language
primer. W3C recommendation, 27(1):123.
Mungall, C. J., Batchelor, C., and Eilbeck, K. (2011). Evo-
lution of the sequence ontology terms and relation-
ships. Journal of biomedical informatics, 44(1):87–
93.
Musen, M. A. et al. (2015). The prot
´
eg
´
e project: a look
back and a look forward. AI matters, 1(4):4.
Noy, N. and Rector, A. (2004). Defining n-ary relations on
the semantic web: Use with individuals. W3C Work-
ing Draft, 21:102.
Noy, N. F., McGuinness, D. L., et al. (2001). Ontology
development 101: A guide to creating your first ontol-
ogy.
Noy, N. F., Shah, N. H., Whetzel, P. L., Dai, B., Dorf, M.,
Griffith, N., Jonquet, C., Rubin, D. L., Storey, M.-A.,
Chute, C. G., et al. (2009). Bioportal: ontologies and
integrated data resources at the click of a mouse. Nu-
cleic acids research, 37(suppl
2):W170–W173.
Overton, J. A., Dietze, H., Essaid, S., Osumi-Sutherland,
D., and Mungall, C. J. (2015). Robot: A command-
line tool for ontology development. In ICBO.
Ptashne, M. (1967). Specific binding of the λ phage repres-
sor to λ dna. Nature, 214(5085):232.
Santos-Zavaleta, A., Salgado, H., Gama-Castro, S.,
S
´
anchez-P
´
erez, M., G
´
omez-Romero, L., Ledezma-
Tejeida, D., Garc
´
ıa-Sotelo, J. S., Alquicira-
Hern
´
andez, K., Mu
˜
niz-Rascado, L. J., Pe
˜
na-Loredo,
P., et al. (2018). Regulondb v 10.5: tackling
challenges to unify classic and high throughput
knowledge of gene regulation in e. coli k-12. Nucleic
acids research, 47(D1):D212–D220.
Sioutos, N., de Coronado, S., Haber, M. W., Hartel,
F. W., Shaiu, W.-L., and Wright, L. W. (2007).
Nci thesaurus: a semantic model integrating cancer-
related clinical and molecular information. Journal of
biomedical informatics, 40(1):30–43.
Smith, B., Ashburner, M., Rosse, C., Bard, J., Bug, W.,
Ceusters, W., Goldberg, L. J., Eilbeck, K., Ireland, A.,
Mungall, C. J., et al. (2007). The obo foundry: coor-
dinated evolution of ontologies to support biomedical
data integration. Nature biotechnology, 25(11):1251.
Towards the Prokaryotic Regulation Ontology: An Ontological Model to Infer Gene Regulation Physiology from Mechanisms in Bacteria
499
