
A NEW LATENT SEMANTIC ANALYSIS BASED METHODOLOGY
FOR KNOWLEDGE EXTRACTION FROM BIOMEDICAL
LITERATURE AND BIOLOGICAL PATHWAYS DATABASES
F. Abate, A. Acquaviva, E. Ficarra and E. Macii
Politecnico di Torino, Turin, Italy
Keywords:
Bioinformatic, Latent semantic analysis, Text mining, Biological pathway.
Abstract:
Nowadays, a considerable amount of genetic and biomedical studies are mostly diffused on the Web and freely
available. This exciting capability, if from one side opens the way to new scenarios of cooperating research,
on the other side makes the knowledge retrieval and extraction an extremely time consuming operation. In this
context, the development of new tools and algorithms to automatically support the scientist activity to achieve
a reliable interpretation of the complex interactions among biological entities is mandatory.
In this paper we present a new methodology aimed at quantifying the biological degree of correlation among
biomedical terms present in literature. The proposed method overcomes the limitation of current tools based on
public literature information only, by exploiting the trustworthy information provided by biological pathways
databases. We demonstrate how to integrate trusted pathway information in a semantic correlation extraction
chain based on UMLS Metathesaurus and relying on PubMed as literature database. The effectiveness of
the obtained results remarks the importance of automatically quantifying the degree of correlation among
biomedical terms in order to helpfully support the scientist research activity.
1 INTRODUCTION
The pace of genetical and biological research has sur-
prisingly spread in the last few years. The devepe-
ment of new technologies (DNA/RNA next gener-
ation sequencing) and the improvement of the old
one (microarray for gene expression at lower costs)
have being supporting the activity of scientists and
researchers. Hundred of experimental results sup-
ported by the biotechnologic enhancement have high-
lighted the mechanisms behind the complex interac-
tions among biological entities as well as brought to
light new unknown phenomena. Nevertheless, if on
one side this rising technological development opens
the way to amazing scenarios in the biomedical re-
search, the problem of handling the great amount of
new information calls for the need of developing new
computational infrastructures. Specifically, the capa-
bility of extracting relevant knowledge from genetical
and biomedical studies has become one of the ma-
jor thrusts in bioinformatic research. Nowadays, the
main sources of information about the biomedical re-
search are mainly biomedical literature databases and
classification systems such as ontologies and thesau-
rus. Most of these systems are distributed on the Web
and thus they are extremely easy to access and opened
to the entire scientific community. However, in this
scenario, the scientist have to face the twofold draw-
back of 1) retrieving the documents containing the in-
formation that he/she is looking for and 2) correlat-
ing the concepts in order to better extract the knowl-
edge they provide. Both the operation are extremely
time-consuming and the degree of correlation among
biological concepts strongly depends on the scien-
tist knowledge background. Therefore, an automated
tool for the computation of a quantified score between
biological terms according to literature information
is strongly necessary in order to support the analysis
of experimental results occurring during the research
activity and to refine the design of the experiments
by focusing on the most relevant features. The cor-
relation score must be the result of a statistical co-
occurrences analysis of biological terms both in the
same article and in the related ones. Moreover, the
tool must be able to integrate as many as possible
sources of information, distributed on the Web and
collected in different formats and structures, in or-
der to guarantee the best accuracy and reliability for
66
Abate F., Acquaviva A., Ficarra E. and Macii E..
A NEW LATENT SEMANTIC ANALYSIS BASED METHODOLOGY FOR KNOWLEDGE EXTRACTION FROM BIOMEDICAL LITERATURE AND
BIOLOGICAL PATHWAYS DATABASES .
DOI: 10.5220/0003171400660074
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2011), pages 66-74
ISBN: 978-989-8425-36-2
Copyright
c
2011 SCITEPRESS (Science and Technology Publications, Lda.)

the computed biological correlation score. Section 2
overviews the state of the art where the proposed tool
is contextualized. Section 3 shows the methodology
used to both retrieve the biological pathway informa-
tion and extract the knowledge by the overall infor-
mation base. Section 4 demonstrates the effectiveness
of the proposed approach in computing the biological
correlation score among biomedical terms. In Section
5 the conclusions and the opening of the future work
are faced.
2 BACKGROUND
To improve the quality of the search over public lit-
erature databases, many search engines have been de-
veloped to classify and correlate the huge amount of
paper that a search may return. Most of this tools
improves the search engine capability adopting on-
tologies and thesaurus as information base. GoP-
ubMed (Doms, A. and Schroeder, M., 2005) is a
search engine tools that make the search on PubMed
(PubMed) repository categorized and ranked. In de-
tails, it applies text-mining algorithm in order to clas-
sify abstracts retrieved from PubMed according to
terms coming from GO and Medical Subject Head-
ings (MeSH) vocabulary (MeSH, 2005). A further
extension to GoPubMed is GoGene (Plake, C. et al.,
2009). The scope of this tool is to rank a list of
genes, occurring in a document set after a PubMed
search, according to terms of GO and MeSH. Af-
ter performing a document search by means of GoP-
ubMed, a text mining algorithm looks for gene names,
GO and MeSH terms over the collection of paper ab-
stracts. Finally, it returns a list of genes associated
with concepts of GO and MeSH. Even if the associ-
ation is performed computing a correlation score be-
tween a single gene and a term, the scope of the score
is not to express a measure of biologic relationship
between either two terms, or two gene, or a gene and
a term, but it is an a posteriori statistical analysis that
allows to map a gene into a ontology term. Further-
more, the problem of computing a quantified semantic
correlation score among biological concepts accord-
ing to the information coming from ontologies has
been addressed in (Wang, J. Z. et al., 2007). In this
work authors developed a tool for measuring the se-
mantic similarity of two Gene Ontology terms (The
Gene Ontology Consortium, 2000). The algorithm
exploits the graph-based hierarchical topology of GO
and it aggregates the semantic contribute of the an-
cestors providing a numeric value that reflects the dis-
tance among GO terms from the biological meaning
point of view. Nevertheless, this tools is limited in
foreclosing a set of biological topics related to the
genetical and biological field and the base of infor-
mation is strongly limited to the GO vocabulary. For
instance, the medical term “Parkinson is not present
in GO, therefore the correlation between this disease
and a certain gene is uncomputable.
A different approach for providing a quantified
measure among biological terms, based on the Vector
Space Model theory is proposed in (Abate, F. et al.,
2010). In this work, the authors apply the Latent
Semantic Indexing (LSI) algorithm (Gliozzo, A. M.
and Strapparava, C., 2005) to a set of document ab-
stracts automatically retrieved through PubMed web
services. The LSI algorithm is strongly based on the
decomposition, by means of Singular Value Decom-
position (SVD) algorithm, of a term-by-document oc-
currences matrix X in three sub-matrix U, Σ and V
where U and V contain the left and right singular
vectors of X and the matrix Σ is a diagonal matrix
containing the singular values. The correlation score
about two terms in the X matrix is calculated by ap-
plying the cosine product of the vector of U-by-Σ ma-
trix corresponding to the correlating terms. Trans-
forming the X matrix into the U-by-Σ matrix allows
to consider not only the occurrences of terms but also
the co-occurrences, exploiting the conceptual links
among terms occurring in the different documents,
but belonging to the same context. The analysis per-
formed by the LSI algorithm is further coupled with a
pre-processing phase based on the Metamap program,
provided by NIH (Aronson, A. R., 2001). The algo-
rithm behind Metamap allows to extract biomedical
concepts from document text so that each abstract is
translated into a list of biomedical concepts, namely a
list of UMLS Concept Unique Identifiers (CUI) (Bo-
denreider, O., 2004). Moreover, the authors empha-
size that the resulting correlation scores, namely Se-
mantic Correlation Score, SRS allow the comparison
among biological concepts belonging to the same re-
trieved document abstract set but it looses accuracy
when comparing terms and concepts belonging to dif-
ferent experimental run and document set. Therefore,
the SRSs go out of the scope of calculating an uni-
versal correlation score as the validity of the compar-
ison is limited to a specific experimental run. In or-
der to overcome this limitation, authors analyze the
density function of the statistical distribution of the
correlation scores within a single experimental run
and divide the density function in percentiles. Con-
sequently, the evaluation through percentiles allow to
evaluate the correlation between genes and biological
concepts independently of the experimental run, be-
cause it provides an information on the frequency dis-
tribution of the SRSs in the overall CUI set occurring
A NEW LATENT SEMANTIC ANALYSIS BASED METHODOLOGY FOR KNOWLEDGE EXTRACTION FROM
BIOMEDICAL LITERATURE AND BIOLOGICAL PATHWAYS DATABASES
67

in an experimental run.
Nevertheless, the effectiveness of the methodol-
ogy introduced in (Abate, F. et al., 2010) strongly de-
pends on the reliance of the information in the re-
trieved abstract documents. Scientific papers, and
specifically those from PubMed as far as the biomedi-
cal field is concerned, are certainly the most consulted
source of information by scientist and biologist be-
cause they contain the more updated state of the art in
science and medicine. However, the nature itself of
biomedical literature is characterized by a significant
level of uncertainty. In fact, the scientific publica-
tions, reporting the state of the art on scientific topics,
are continually evolving and, as a consequence, the
addicted argumentations might be often contradict-
ing. Therefore, a biological correlation score com-
puted by considering documents coming exclusively
from the biomedical literature is affected by a certain
level of uncertainty. For this reason, integrating the
set of documents coming from literature with trusted
information is a mandatory step in order to enhance
the computational accuracy of the biological correla-
tion among biological concepts. In order to get the
scope, a twofold question must be answered: firstly,
what are the trusted source of informations and to
what extend a source of information can be consid-
ered as trusted; secondly, once the trusted informa-
tion have been collected, what is the more effective
way to integrate this information in the Latent Seman-
tic Analysis algorithmic infrastructure?
2.1 Trusted Information Sources
It is worth underlining that several sources of biomed-
ical information, mainly ontologies and thesaurus, are
universally accepted as trusted source of information.
GO, for instance, correlates gene and gene products
by means of annotations. GO curators coordinate
the development of the ontology rooted in the exper-
imental literature through an annotation-driven pro-
cess and, moreover, the accuracy of the relations and
terms definitions are continually checked (Hill, D. P.
et al., 2008). Moreover, genetic and metabolic path-
ways are a graph-based representation of the interac-
tion of genes and proteins in a biological process.
The annotations related to new discovered path-
ways are collected in several pathway databases.
These databases play a key role in the genetic and bio-
logic research field. Examples of pathways databases
that have been developed in the last years are the Ky-
oto Encyclopedia of Genes and Genomes (KEGG)
(Kanehisa, M. and Goto, S., 1999), BioGRID (Stark,
C. et al., 2006), HumanCyc (Romero, P. et al., 2004).
On the base of pathways information, a number of
knowledge based tools have been developed. In par-
ticular, Pathway Commons is a projects that aims
at integrating all the distributed databases and the
pathway information under a common web interface
(Pathway Commons, 2007), providing a single ac-
cess point to multiple pathway data sources. Path-
way Commons runs on cPath software engine (Ce-
rami, E. G. et al., 2006), an open source framework
that makes the aggregation of custom pathway data
sets easy. The key feature of cPath is mainly the
ability to support standard pathways exchange format
as PSI-MI (Hermjakob, H. et al., 2004) and BioPax
(BioPAX, 2007), as well as providing all the informa-
tion content in XML format through web service API.
Moreover, cPath stores external link records matching
the biological entity with Gene Ontology terms if the
references are present in the PSI-MI file.
2.2 Integration of Trusted Information
in the LSI Algorithm
LSI is particularly effective for text clustering ap-
plications because it exploits co-occurrences among
terms in order to gather latent associations (Gliozzo,
A. M. and Strapparava, C., 2005). This feature makes
the LSI particularly successful in handling synonyms
and polysemys. However, LSI considers all the words
in the documents set as a bag of words, where all the
terms are equally weighted. In (Chakraborti, S. et al.,
Figure 1: Modified Occurrences Matrix with sprinkling la-
bel classes.
2006), authors introduce the “sprinkling”, an efficient
technique that enhances text classification accuracy
taking into account document class labels. Label-
ing the documents helps LSI promoting inferred la-
tent associations between words conceptually belong-
ing to the same class. Considering the LSI algorithm,
the documents labeling results in creating a new oc-
currences matrix where new terms are added. Fig-
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
68
ure 1 shows how sprinkling technique influences the
occurrence matrix content during the LSI algorithm
computation. The class-labelled terms corresponds
to extra rows with non-zero value only in the la-
belled documents, and all-zeros otherwise. The term
rows added by the sprinkling artificially create new
co-occurrences among terms belonging to the same
document class making explicit the implicit associa-
tions. Therefore, the efficiency in promoting implicit
associations makes the sprinkling process a good can-
didate to integrate trusted informationin the LSI algo-
rithm in order to promote implicit associations among
terms embedded in the document set. Starting from
LSI and the sprinkling idea we developed a new cus-
tomized methodology in order to integrate the asso-
ciations coming from the trusted biological pathway
with the information included in a set of documents
retrieved from PubMed web services.
3 METHODOLOGY
The complete flow of the propose method is mainly
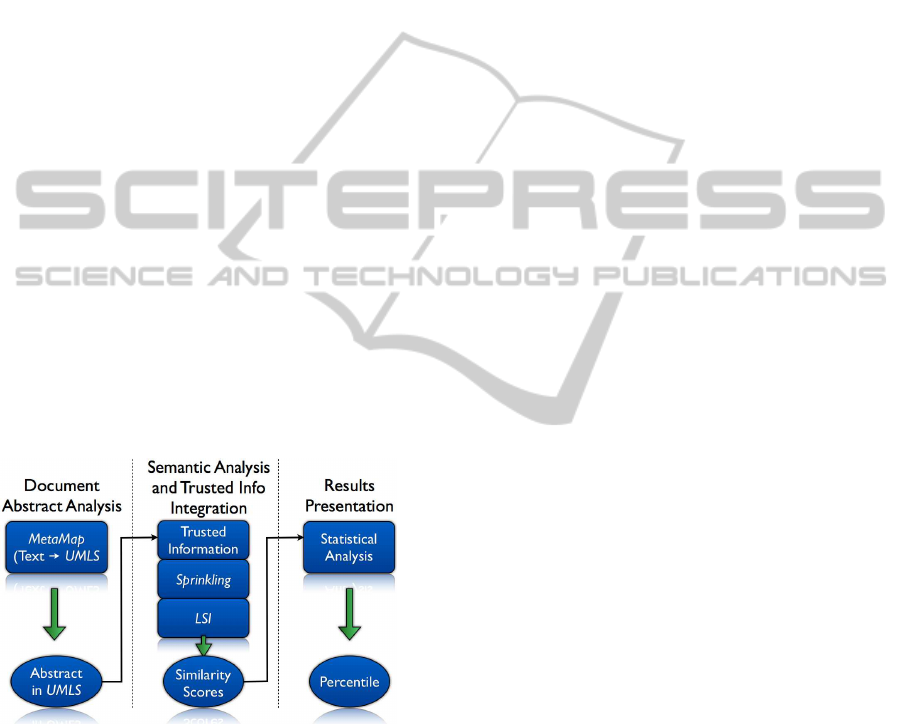
composed by three phases depicted in Figure 2:
Document Abstract Analysis, Semantic Analysis and
Trusted Info Integration, Results Presentation. The
following sub-sections describe the methodologies
behind each phases in more details.
Figure 2: Complete Semantic Analysis Flow.
3.1 The Document Abstract Analysis
Phase
The initial set of documents is composed by ab-
stract retrieved through PubMed web services. In
particular, we consider the case the scientist is in-
terest in quantify the biological correlation score
among a biological process and a specified gene.
The set of keyword used to query the PubMed web-
service is composed by the biological process and the
gene name (initial keyword set), and a list of syn-
onyms and terms correlated to both the gene and the
biological process (expanded keyword set), resulting
from querying the UMLS database(Bodenreider, O.,
2004). In detail, in order to get the more complete
set of synonyms and correlated terms, we exploit the
fact that ULMS database store the relations among
concepts belonging to almost 37 different biomedi-
cal database and ontologies (Bodenreider, O., 2004).
Hereafter, we select the relevant relations in order to
download the more related abstract set, exploiting the
methodology introduced in (Abate, F. et al., 2010). It
is worth noting that the more the retrieved abstracts
relate both the biological process and the gene, the
more the resulting biological score is accurate. In fact,
the presence of unrelated with the specified terms in-
creases the noising information that in turn alter the
result accuracy.
Moreover, in the Document Abstract Analysis
phase, the retrieved set of document abstracts from
PubMed written in Natural Languageare translated in
a Concept Language exploiting Metamap capability.
In fact, Metamap allows to map natural language text
in Concept Unique Identifiers (CUI). Pre-processing
the abstracts by means of Metamap makes the seman-
tic analysis more accurate in that it reduces the am-
biguities among terms that express the same concept
with different sentences and words, and it allows to
reduce non-relevant terms filtered by Semantic Type
(Abate, F. et al., 2010). Therefore, at the end of
Document Abstract Analysis phase, the set of abstract
documents from PubMed translated in Concept Lan-
guage is returned forming the set of documents on
whom the semantic analysis is performed.
3.2 The Semantic Analysis and Trusted
Info Integration Phase
This phase is the main core of the proposed flow and
it consists of 1) retrieving the Trusted Information, 2)
applying the sprinkling technique in order to integrate
the information from the document abstract, 3) per-
forming the latent semantic analysis in order to get
the most accurate SRS list .
3.2.1 Trusted Information
The proposed tool considers genetical pathways as
trusted source information. In fact, genetical path-
ways are a graphical-based representation of the gene
involved in a specific biological pathways. Thus, they
provide a direct link between a gene and the corre-
sponding biological process and in this sense they are
good candidate as additional source of information
A NEW LATENT SEMANTIC ANALYSIS BASED METHODOLOGY FOR KNOWLEDGE EXTRACTION FROM
BIOMEDICAL LITERATURE AND BIOLOGICAL PATHWAYS DATABASES
69

Figure 3: GetPathwayInformationByBiologicalProcess pro-
cedure to get trusted information.
to the biomedical abstracts set. Moreover, genetical
pathways are constantly subjected to a review process
and consequently they can be considered as trusted.
In order to automatically access genetical pathways
information, a software client for querying Pathway
Commons web-service has been developed. In
details, pathways information are extracted through
the GetPathwayInformationByBiologicalProcess
procedure shown in Figure 3.
GetPathwayInformationByBiologicalProcess re-
turns from Pathway Commons the list of pathways
where the biological process specified as input occurs
in the pathway description or in the labeling title.
Hereafter, for each pathway in the list, the cpath id
(i.e. the unique pathway identifier according to cPath
framework (Cerami, E. G. et al., 2006), is extracted
from the pathway description and it is used to access
the complete set of genes involved in the pathway.
It is worth noting that the presented method
achieves a twofold scope: firstly, the link between
the specified gene and biological process is con-
firmed consulting a trusted source of information;
secondly, a list of gene trustly involved in the spec-
ified biological process are collected. The latter in-
formation is particularly tailored in enhancing the co-
occurrences of genes correlated with the specified
gene and biological process, increasing the overall ac-
curacy during the semantic analysis.
3.2.2 Applying Sprinkling to Semantic Analysis
In this phase, the information extracted from pathway
database are integrated in the semantic analysis pro-
cess of PubMed abstracts. The term-by-document oc-
currence matrix is the main data structure processed
by the LSI algorithm and, consequently, it is the
main access point to integrate additional information.
The term-by-document occurrence matrix is a two-
dimensional occurrence matrix in which the columns
represent the dimensional space of a corpus of docu-
ments and the rows represent the dimensionalspace of
Figure 4: Occurrences Matrix adding pathway informa-
tion. The dashed line separate the abstract document set
by document pathways.
the terms occurring into the documents. However, the
abstract set and the retrieved pathways present a dif-
ferent structure of the informational content. The ab-
stract set is composed by documents already suitable
for the latent semantic analysis algorithm, whereas
the pathways are represented as a connected graph.
Therefore, in order to integrate pathways content in
the semantic analysis flow, the information coming
from pathways are firstly extracted and translated into
a new document containing the trusted information.
In detail, the created trusted document contains the
list of gene names involved in the retrieved path-
ways as well as the list of retrieved biological path-
ways. Nevertheless, adding a single document to
the overall document set weakly affects the seman-
tic analysis in terms of accuracy because poorly rel-
evant from the statistical point of view. Thus, the
trusted document, containing the extracted pathway
information, is replicated. We refer at this procedure
as document padding. Figure 4 shows a trivial term-
by-document occurrences matrix sprinkled with addi-
tional document padding. The number of instances of
the padded document, namely NPD, is obtained ac-
cording the following equation:
NPD = ⌈p· NAD⌉ (1)
where NAD is the total number of PubMed abstract
document and p is a padding factor spanning from
0 to 1. Consistently with this equation, the num-
ber of padded document spans from zero to the same
number of document abstracts. Padding the overall
document set with trusted information affects the sin-
gular values of the occurrence matrix. In fact, the sin-
gular values reflect on the distribution of the prevalent
concepts occurring in the document set. The higher
the singular value score, the greater is the presence
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
70
of the concept in the overall document corpus. It is
worth noting that increasing the p factor makes the
trusted concepts included in the trusted documents
predominant with respect to the overall concepts.
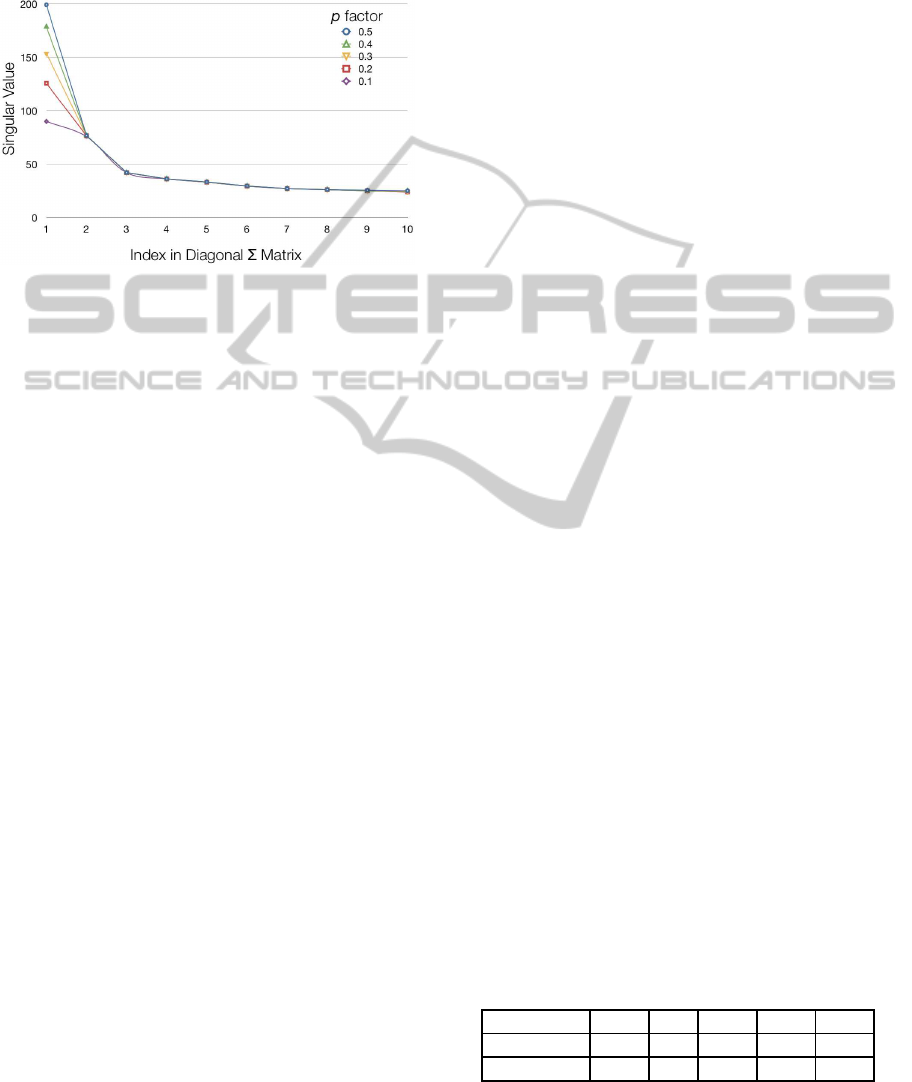
Figure 5: Singular values obtained launching the latent se-
mantic analysis for computing the biological correlation
score between AKT1 gene and Angiogenesis biological
process. The ordinate reports the singular value score and
the abscissa represents the index in the diagonal singular
value matrix.
The analysis of the singular values is fundamental
to evaluate how the trusted document padding affects
on the overall document corpus by coherently tuning
the p factor value. In the LSI algorithm, the singular
values, obtained after applying the SVD, represent the
attendance of the concepts in the analyzed documents
set. The more a concept is relevant and frequent in
the document set the higher is the singular value cor-
responding to it. Moreover,during the latent semantic
analysis the singular values computed by SVD are set
in the diagonal Σ matrix in ascending order, and con-
sequently the first singular values represent the more
relevant concepts in the analyzed corpus. Figure 5
plots the first ten singular values resulting from the
analysis of abstract document set for the computa-
tion of the correlation score among the Angiogenesis
biological process and AKT1 gene term. The singular
values are obtained with five different p factor values.
When the document padding is increased by tuning
the p factor, the concepts in the trusted documents be-
come more relevant respect to the concepts in the ab-
stract document set. When the p factor increases even
more the concepts in the trusted document set become
so relevant that the concepts in the abstract document
set are overcome. As a consequence, the singular
values corresponding to the trusted document set are
boosted up and they occupy the first positions in the
diagonal Σ matrix. This is the reason why, as shown
in Figure 5, the greater the p factor value, the higher
the first singular value score. Therefore, with greater
p factor values the most relevant concepts in the over-
all document set correspond mainly to the concepts in
the trusted document set.
The alteration on the singular values of the occur-
rence matrix by the trusted document padding con-
sequently reflects on the correlation score between
the biological terms. Consider the scenario depicted
in Figure 4: TermA and TermB present a weak cor-
relation in the document abstract set because quite
linearly independent, whereas they are strongly cor-
related in the trusted document set. The document
padding increases the parallelism of the two terms
vector, consequently enhancing the correlation score.
Furthermore, both TermA and TermB are correlated
with TermC creating an indirect correlation due to co-
occurrence. If the TermC occurs even in the trusted
document set the resulting correlation score between
TermA and TermB is further enhanced. Moreover,
when a term occurs both in the document abstract
set and in the trusted documents set, the term con-
ceptually belongs to the trusted concepts as well. If
two biological terms belong to the trusted concepts
their correlation score is higher because conceptu-
ally linked to the predominant concepts in the over-
all documents content. Specifically, the biological
correlation score between the biological process and
the gene term is influenced by trusted information
by a twofold effect: in the case the terms corre-
sponding to biological process and gene term occur
both in the document abstract set and in the trusted
documents the score is directly affected; moreover,
the correlation score is further enhanced by the pres-
ence of terms correlated with both the biological pro-
cess and gene terms because occurring both in the
document abstract set and in the trusted document
set. Table 1 shows the correlation scores between
Angiogenesis and AKT1 gene, both in SRSs and in
Percentile. The reported results show that increasing
the p factor value implies a corresponding increase in
the correlation score. Therefore, it is possible to assert
that the proposed methodology, provides an effective
correlation score combining the information extracted
in the document abstract set with the trusted informa-
tion extracted from pathways databases. Moreover,
the influence of the trusted information on the seman-
tic text analysis is tuned by the p factor parameter that
directly affects the correlation results.
Table 1: SRS and Percentile values corresponding to p fac-
tor.
p Factor 0.1 0.2 0.3 0.4 0.5
SRS 15.7 17 18.1 18.8 19.5
Percentile 76 78 82 83 84
A NEW LATENT SEMANTIC ANALYSIS BASED METHODOLOGY FOR KNOWLEDGE EXTRACTION FROM
BIOMEDICAL LITERATURE AND BIOLOGICAL PATHWAYS DATABASES
71
3.3 The Results Presentation Phase
An evaluation criterion allowing the comparison
among each gene in the genes set and each biological
process is fundamental in order to focus the atten-
tion only on those genescorrelated with the biological
processes of interest behind a certain degree of cor-
relation. Therefore, the correlation measurement
between genes and biological processes must ex-
press an universally valid score allowing the compar-
ison among different correlation analysis. Actually,
SRS provides a percentage measurement to compare
genes and biological process corresponding to terms
belonging to the same occurrence matrix and thus
it quantifies the semantic relationship of biological
terms during the same semantic text analysis (i.e.
a single execution of the tool). Different analyses
are characterized by different sets of documents and
concepts. Consequently, SRSs values computed on
different occurrences matrices are not directly compa-
rable. In order to define an evaluation criterion that al-
lows to compare SRSs resulting from differentseman-
tic analysis, and therefore different occurrences ma-
trices, we analyzed the SRS distribution on different
occurrences matrix. The SRS Distribution (SRSD) is
defined as the density function that reports the per-
centage of occurrences, or frequencies, of a certain
score for the possible SRSs corresponding to all the
CUIs occurring within the correlation analysis. Fur-
thermore, the SRSD has been divided into hundred
percentile intervals of equivalent area depending on
the frequency percentage of the SRS in the density
function. We define each interval as Biological Cor-
relation Index (BCI) expressing that the higher the
BCI, the greater the degree of biological correlation
between the biological process and the genes. The
evaluation through BCI allows to evaluate the corre-
lation between genes and biological process indepen-
dently of the experimental run, because it provides an
information on the frequency distribution of the SRSs
in the overall terms occurring in an experimental run.
4 EXPERIMENTAL RESULTS
The following section reports the correlation
scores among a set of three biological processes
(Angiogenesis, Apoptosis, and Signal Transduction)
and ten gene terms (VEGFA, ANGPT2, ANGPT1,
AKT1, BCL-xL, BCL-2, P53, PTEN, MAPK1,
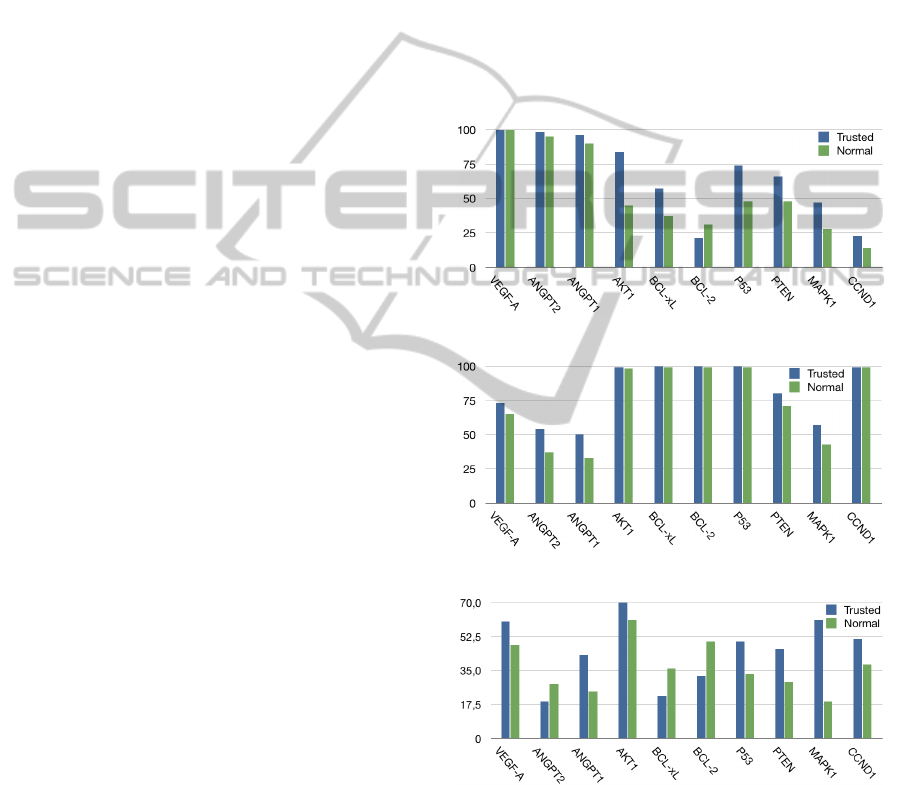
CCND1) applying the proposed flow. Figure 6 re-
sumes the results and demonstrate the effectiveness of
integrating the information coming from the PubMed
document abstract set with the information from
pathway database. The biological correlation scores
obtained integrating trusted information (Trusted
INFO) are compared with the scores obtained without
integrating any trusted information (Normal INFO).
Moreover, p factor has been set to 0.5. This choice
implies that the trusted document set has been padded
in order to equal almost the half of the number of
abstract document set and to emphasize the effect of
the integration of trusted information on the overall
document set. Furthermore, the biological correlation
score is reported in the BCI format in order to allow
the comparison among the scores of same set of genes
computed in different biological process (Abate, F.
et al., 2010).
(a) Angiogenesis.
(b) Apoptosis.
(c) Signal Transduction.
Figure 6: Experimental Results Expressed in BCI score.
In order to have a better understanding of the re-
sulting biological correlation scores, the functional
definition of the gene set according to NCI Thesaurus
is listed below:
• VEGFA: This gene is involved in the regulation of
blood vessel growth.
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
72

• ANGPT2: This gene encodes angiopoietin-2
protein and it plays a role in both angiogenesis
and neovascularization.
• ANGPT1: This gene plays a role in both vascular
development and angiogenesis.
• AKT1: This gene is involved in signal transduc-
tion and negative regulation of apoptosis.
• BCL-xL: This protein plays a role in the modula-
tion of apoptosis.
• BCL2: This gene is involved in apoptotic regu-
lation. Overexpression of this gene promotes the
pathogenesis of B-Cell lymphomas, due to anti-
apoptotic activity.
• P53: This protein plays a role in the regulation of
both the cell cycle and apoptosis.
• PTEN: This gene plays a role in signal transduc-
tion and apoptosis. It is also involved in the regu-
lation of cell cycle progression.
• MAPK1: This gene plays a role in signal trans-
duction and positive regulation of the cell cycle.
• CCND1: This gene plays a role in the regulation
of mitotic events.
From the same NCI Thesaurus source,
Angiogenesis is defined as the “blood vessel
formation” and, specifically for the case of tumor
angiogenesis, it is “the growth of blood vessels from
surrounding tissue to a solid tumor”. It is worth
noting that the results concerning the biological
correlation scores between angiogenesis and the gene
set coherently reflect the NCI Thesaurus definitions.
In fact, VEGFA, ANGPT1 and ANGPT2 present the
almost same correlation scores with the Angiogenesis
biological process both in the case of Trusted INFO
and in the case of Normal INFO. The small difference
in the results depends on the fact that in both the cases
the biological correlation scores presents highest
BCI, therefore the Trusted INFO poorly affect the
accuracy of the computation. Moreover, the corre-
lation score of AKT1 gene term with Angiogenesis
process is highly enhanced by the integration of
Trusted INFO. This gene is specifically involved in
the Signal Transduction and in the Apoptosis process,
whereas it is less typical of Angiogenesis biological
process respect to VEGFA, ANGPT1, and ANGPT2
genes even if correlated with it. Our results reflects
this assertion because AKT1 gene directly appears in
the Angiogenesis pathway and it is directly correlated
with this biological process according to the trusted
information set, but its correlation according to the
information coming from the document abstract
set is minor compared with VEGFA, ANGPT1, and
ANGPT2 genes. Coherently, this gene presents a
lower biological correlation score compared with
VEGFA, ANGPT1, and ANGPT2 genes even if,
correctly, its score has been significantly boosted by
adding Trusted INFO.
Moreover, the integration of Trusted INFO en-
hances the biological correlation score of P53, PTEN,
MAPK1 and CCND1. These genes are generally in-
volved in Apoptosis, Signal Transduction and Cell
Cycle biological processes that are indirectly related
with Angiogenesis process as well. The AKT1 gene,
that is mainly involved in Signal Transduction and
Apoptosis, also occurs in the Angiogenesis pathway.
The biological correlation of the AKT1 gene with
P53, PTEN, MAPK1 and CCND1 genes and, on the
other side, the occurrence of this gene in the trusted
document concerning Angiogenesis process creates
an indirect link between P53, PTEN, MAPK1 and
CCND1 genes and the same Angiogenesis process. It
accordingly results in a biological correlation score
enhancement.
Furthermore, the introduction of Trusted infor-
mation positively increases ANGPT1, ANGPT2 and
MAPK1 genes scores. These genes are generally typi-
cal of Angiogenesis and Signal Transduction but these
processes are sometime related to Apoptosis and thus
they co-occur in the document abstact set. There-
fore, even if the Apoptosis pathway do not include this
gene set, the indirect biological correlation between
terms in the Trusted information and terms occurring
in the document abstract set, belonging to the Apop-
tosis semantic area, increases the resulting value of
the biological correlation score. However, all of them
present a score significatively lower than the one of
Apoptosis genes AKT1, BCL-xL, BCL2, and P53.
According to the results concerning Signal Trans-
duction process, the biological correlation score of
MAPK1 gene is particularly interesting. This gene
is strongly related to Signal Transduction, but this
biological processes is quite generic and it interact
with many biological phenomena typical of the cell
cycle. Considering only the document abstract set
as information base, the occurrence of MAPK1 gene
is not always frequently guaranteed, thus resulting
in a low correlation score. However, integrating the
Trusted information the score is almost triplicated.
The introduction of Trusted information creates the
missing conceptual links of the MAPK1 gene with
terms semantically correlated with the biological pro-
cess of interest, resulting in an increase of the final
biological correlation score. This result remarks the
effectiveness of the proposed approach in enhancing
accuracy of measuring the biological degree of corre-
lation.
A NEW LATENT SEMANTIC ANALYSIS BASED METHODOLOGY FOR KNOWLEDGE EXTRACTION FROM
BIOMEDICAL LITERATURE AND BIOLOGICAL PATHWAYS DATABASES
73

Furthermore, integrating Trusted information in the
document abstract set makes the results more accurate
also reducing the biological correlation score. BCL2
gene, for instance, is functionally specific of Apopto-
sis biological process. On the wave of this assertion,
this gene further decreases the correlation score in the
case of both Angiogenesis and Signal Transduction
when applying Trusted information. The expected be-
havior would be at most of unaltered result because
the Trusted information do not addict any further in-
formation concerning the correlation score between
BCL2 and the two biological processes. However,
integrating Trusted information increases the corre-
lation score of those terms related to the biological
processes and their correlation scores overcomes the
BCL2 one, that in turn becomes lower. Similar con-
sideration are also valid about the biological correla-
tion score between BCL-xL gene and ANGPT2 gene
and Signal Transduction biological process.
5 CONCLUSIONS
The proposed semantic analysis tool provides a
framework for measuring the biological correlation
score among biomedical terms. The score is the
results of a text mining analysis performed on the
abstracts of the most relevant scientific publications
with the support of the completeness of the UMLS
Metathesaurus. The knowledgeextracted by biomedi-
cal literature is further integrated with the information
coming from sources generally known as trustworthy.
In order to satisfy this requirement, public biological
pathways databases have been chosen and the embed-
ded information are first retrieved and then correlated
in the overall semantic analysis flow. Moreover, in
order to integrate the knowledge coming from an het-
erogeneous base of information, a new version of the
latent semantic analysis, based on the sprinkling tech-
nique, has been developed. The results remark the
efficiency of the proposed approach in enhancing the
accuracy of the biological correlation score computa-
tion by combining the knowledge extracted by scien-
tific document abstract set with the pathways infor-
mation.
As future step the complete automation of the tool
will allow to retrieve the document abstract set in an
automated and accurate manner. Moreover, the in-
tegration of information coming from other trusted
sources will be tailored and developed in order to bet-
ter improve the overall accuracy and robustness of the
proposed semantic analysis method.
REFERENCES
Abate, F., Ficarra, E., Acquaviva, A., and Macii, E. (2010).
An automated tool for scoring biomedical terms cor-
relation based on semantic analysis. In International
Conference on Complex, Intelligent and Software In-
tensive Systems.
Aronson, A. R. (2001). Effective mapping of biomedical
text to the umls. metathesaurus: The metamap pro-
gram. In AMIA Fall Symposium.
BioPAX (2007). Biological pathways exchange.
http://www.biopax.org.
Bodenreider, O. (2004). The unified medical language sys-
tem (umls): integrating biomedical terminology. Nu-
cleic Acids Research.
Cerami, E. G., Bader, G. D., Gross, B. E., and Sander, C.
(2006). cpath: open source software for collecting,
storing, and querying biological pathways. In Bioin-
formatics.
Chakraborti, S., Mukras, R., Lothian, R., Wiratunga, N.,
Watt, S., and Harper, D. (2006). Sprinkling: Super-
vised latent semantic indexing. Advances in Informa-
tion Retrieval.
Doms, A. and Schroeder, M. (2005). Gopubmed: explor-
ing pubmed with the gene ontology. Nucleic Acids
Research.
Gliozzo, A. M. and Strapparava, C. (2005.). Domain ker-
nels for text categorization. In Ninth Conference on
Computational Natural Language Learning.
Hermjakob, H. et al. (2004). The hupo psi’s molecular inter-
action format–a commu- nity standard for the repre-
sentation of protein interaction data. Natural Biotech-
nology.
Hill, D. P., Smith, B., McAndrews-Hill, M. S., and Blake,
J. A. (2008). Gene ontology annotations: what they
mean and where they come from. In Bioinformatics.
Kanehisa, M. and Goto, S. (1999). Kegg: Kyoto encyclo-
pedia of genes and genomes. Nucleic Acids Research.
MeSH (2005). Medical subject headings (mesh) fact sheet.
National Library of Medicine.
Pathway Commons (2007). Pathway commons.
http://www.pathwaycommons.org.
Plake, C., Royer, L., Winnenburg, R., Hakenberg, J., and
Schroeder, M. (2009). Gogene: gene annotation in
the fast lane. Nucleic Acids Research.
Romero, P., Wagg, J., Green, M. L., Kaiser, D., Krummen-
acker, M., and Karp, P. D. (2004). Computational pre-
diction of human metabolic pathways from the com-
plete human genome. Genome Biology.
Stark, C., Breitkreutz, B. J., Reguly, T., Boucher, L., Bre-
itkreutz, A., and Tyers, M. (2006). Biogrid: a general
repository for interaction datasets. Nucleic Acids Re-
search.
The Gene Ontology Consortium (2000). Gene ontology:
tool for the unification of biology. Nature Genetics.
Wang, J. Z., Du, Z., Payattakool, R., Yu, P. S., and Chen,
C. F. (2007). A new method to measure the semantic
similarity of go terms. In Bioinformatics.
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
74
