
GAST, A GENOMIC ALIGNMENT SEARCH TOOL
Kalle Karhu, Juho M¨akinen, Jussi Rautio, Jorma Tarhio
Department of Computer Science and Engineering, Aalto University, Espoo, Finland
Hugh Salamon
AbaSci, LLC, San Francisco, U.S.A.
Keywords:
Sequence alignment, Text algorithms, Indexing.
Abstract:
Alignment to a genomic sequence is a common task in modern bioinformatics. By improving the methods
used, significant amount of time and resources can be saved. We have developed a new genomic alignment
search tool, called GAST, for sequences of at least 160 nt. GAST is many times faster than commonly used
alignment tools BLAT and Mega BLAST. As the sizes of query sequences and the database increase, the
advantage grows. This paper describes the principles of GAST and reports a comparison of GAST with BLAT
and Mega BLAST. The effects the query sequence length and the number of queries have on run times were
studied using the full human genome and the chromosome 1 of human genome separately. Additionally, the
error tolerance and behaviour of GAST when handling sequences with lower similarity to a database was
studied. Lastly, we compared the quality of exon mappings produced by the three tools and the genomic
mapping tool GMAP.
1 INTRODUCTION
Constructing an alignment of query sequences to a ge-
nomic sequence is a common task in modern bioin-
formatics. NCBI BLAST (NCBI, 2009) alone re-
ceives over 100,000 alignment queries a day. The
computational requirements of these searches amount
to a notable use of resources. Although sequence
alignment has been extensively studied, there remains
room for improvement in the speed of alignment and
the reliability of mapping query sequences with dif-
ferences from the database sequences. In this paper,
the terms query sequence, query, and pattern will be
used interchangeably to stand for the sequence the
user wishes to align or map to database text sequence
or sequences. Concerning the speed, two very popu-
lar alignment methods, Mega BLAST (Zhang et al.,
2000) and BLAT (Kent, 2002), stand out. Both are
similar to the BLAST basic local alignment search
tool (Altschul et al., 1990) in many ways.
Mega BLAST’s performance is increased by us-
ing a “greedy algorithm”, which starts three different
lines of further processing whenever an error is en-
countered. These three lines correspond to (i) han-
dling a mismatch, (ii) an insertion in the query, and
a deletion in the query. When a difference between
the query and the database occurs, one of the lines is
likely to continue running as the other two will termi-
nate immediately. With high similarity between the
query and the database, this method is computation-
ally very effective.
BLAT uses indexing of all suitably sized, non-
overlapping k-mers in the database. The index is used
in a search phase to connect these k-mers to the k-
mers of the query sequence. Lastly, an alignment is
done by extending the sites found in the search phase.
In this paper we present an alignment method
which is considerably faster than the aforementioned
methods Mega BLAST and BLAT. The phase struc-
ture of our method is similar to that of BLAT: index-
ing, searching, and alignment. However, the logic of
each phase in our tool is different from BLAT. The
indexing phase in our method collects all k-mers of
fixed length following dinucleotides AC, which we
call AC-probes. Reasoning behind this kind of k-mer
selection, together with the choice of dinucleotide
AC out of all dinucleotides, is explained in Section
2.1. Information describing the approximate sites of
the occurrences of these AC-probes in the database
is saved. The search phase compares the AC-probes
82
Karhu K., Mäkinen J., Rautio J., Tarhio J. and Salamon H..
GAST, A GENOMIC ALIGNMENT SEARCH TOOL.
DOI: 10.5220/0003181400820090
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2011), pages 82-90
ISBN: 978-989-8425-36-2
Copyright
c
2011 SCITEPRESS (Science and Technology Publications, Lda.)

found in the pattern to the ones in the database se-
quence. If enough AC-probes from the pattern are
found from a certain section of the database, this sec-
tion is selected for further processing. Finally, the
alignment phase further refines mapping. This phase
starts with the BG algorithm (Salmela et al., 2006),
which searches for candidates of alignment locations.
Our tool GAST (short for Genomic Alignment
Search Tool), yields results many times faster than
the aforementioned BLAT and Mega BLAST. As
the sizes of queries and the databases increase, our
method outperforms the aforementioned methods by
greater margins. Common tasks involving relatively
long patterns include mapping of cDNA and ex-
pressed sequence tags to genomes. Additionally
GAST has proved to be more tolerant to errors or
small differences between the database text and the
pattern, and thus was able to map such query pat-
terns faster and more reliably than BLAT and Mega
BLAST.
Originally GAST was made with sequences of
length 1000 nt and above in mind. Later on we dis-
covered that GAST is able to provide reliable results
with a lot shorter sequences too and the lower border
can be further on adjusted by changing some of the
parameter values. With this in mind, we compared
the quality of the exon mappings generated by GAST,
Mega BLAST and BLAT. Additionally, we compared
the results to a fourth tool, GMAP (Wu and Watan-
abe, 2005), designed specifically for cDNA mapping.
GMAP performs genomic mapping using rela-
tively long, 24 nt oligomers. This mapping requires
the usage of an index file, similarly to BLAT and
GAST. The results of the initial mapping are further
refined using oligomer chaining and a specific type of
dynamic programming, called sandwich DP.
Another tool which is close to the mentioned tools
in performance is SSAHA (Ning et al., 2001). Ac-
cording to a comparison published in (Harper et al.,
2006), the quality of the results achieved by SSAHA
is on par with that of BLAT and Mega BLAST. We de-
cided it would not be necessary to include SSAHA in
our comparison, as there is no remarkable difference
in its performance compared to the other two tools.
2 METHODS
Our method can be divided into three different phases:
the creation of a block-addressingq-sample index, the
initial search phase, and lastly the alignment phase,
where the results of the initial search phase are pro-
cessed in a greater detail. The index phase is a prepro-
cessing step, which has to be done only once for each
genome or other collection of database sequences.
Initial search phase uses the index created to find po-
tential sites with high probability of leading to a good
alignment. The alignment phase performs a more pre-
cise alignment between these sites and the patterns
provided. The workflow of our tool in these three
phases is illustrated in Figure 1. The nature of our
tool is relatively heuristic and there are many param-
eters and thresholds involved. These parameters and
choices behind their values are more thoroughly dis-
cussed in Section 2.4.
2.1 Block-addressing Q-sample Index
Our tool uses an index file to gain speed-up in the ini-
tial, approximate search. Essentially, this index struc-
ture combines q-sample filtration (Sutinen and Tarhio,
1996) with block-addressing(Manber and Wu, 1994).
The workflow of our index structure, which is de-
scribed in this subsection, is also visualized in left
section of Figure 1.
The index structure is formed as follows. Given
database files containing the database sequences are
initially divided into blocks of given size b. The file-
names the blocks correspond to and the starting po-
sitions of the blocks in the files are saved in order to
access the sequences of each block. A unique block
ID number is given to each block to act as a key to
this information.
Following the division to blocks, the database se-
quences are scanned for occurrences of a certain din-
ucleotide, AC. These dinucleotide occurrences are ex-
panded to what we call AC-probes. This expansion is
done by taking the 10 nucleotides following the din-
ucleotides AC, resulting in 12-mers. The blockwise
locations of these probes are initially collected. Af-
ter all the blocks of the database sequences have been
scanned, the most frequently occurringAC-probes are
discarded. This censoring is done to reduce the num-
ber of blocks and probes to be processed. The portion
of AC-probes discarded is controlled by ACdisc pa-
rameter. The remaining AC-probes will be referred to
as approved AC-probes.
Resulting from these phases, the index holds a list
of block ID numbers for the collection of approved
AC-probes. Using this index structure, our tool can
rapidly retrieve blocks with occurrences of a given
AC-probe, or a collection of multiple AC-probes.
When creating such block-addressing index based
on oligomer positions, the oligomers used should
be infrequent enough to create distinction between
blocks. Longer oligomers are more infrequent, but re-
sult in larger and computationally heavier index struc-
tures, if occurrences of all oligomers of chosen length
GAST, A GENOMIC ALIGNMENT SEARCH TOOL
83

are indexed. Because of this tradeoff, it is advanta-
geous to have a way of choosing certain portion of
the longer oligomers, in order to cut down the size of
the index. Lastly, this portion of the oligomers should
be easily and rapidly detectable, when scanning the
database or the pattern. The AC-probe satisfies these
requirements and thus it was chosen to be the base
structure of our index. The choice of dinucleotide
AC out of all dinucleotides is supported by (Zhang
and Yang, 2005), (Zhang and Yang, 2008) and (Kar-
lin and Burge, 1995), showing a good combination of
low mean and low variance of incidence for the dinu-
cleotide AC in bacteria, archae and eukaryotes alike.
There is also another preprocessing step required
to be done once for each database file. This is the k-
mer encoding of the text, used by the alignment phase
of our tool. By default, we use the value of q = 7.
The encoded databases are saved in a particular binary
format instead of the ASCII format used in FASTA
files.
2.2 Initial Search
The initial search phase essentially compares AC-
probe profiles of database blocks, which were re-
trieved in the indexing phase, to the AC-probe pro-
files of patterns. As the output of this initial search,
our tool gives the blocks having high probability of
containing a hit for a pattern. Workflow of the initial
search is pictured in the mid section of Figure 1.
Going phase by phase, the initial search is done
as follows. First, the occurrences of AC-probes in the
query sequence are collected. Using these, the sum of
approved AC-probes matching between each pattern
and each block are calculated.
There are two separate threshold values control-
ling the selections of blocks for further processing.
Each block is required to contain at least a portion mp
of approved AC-probes found in the query. The sec-
ond threshold requires each block to contain at least
a portion rp of maximum amount of approved AC-
probes matching between a block and the query se-
quence. If two neighboring blocks do not overcome
the thresholds as solo, but do so when combined, they
are combined and handled as a single block.
If wished so by the user, the search can be stopped
here and the blocks which overcame the thresholds
will be reported with the amount of approved AC-
probes found from them. Otherwise, the search will
be further refined in alignment phase.
2.3 Alignment
The last refining phase in our tool is the alignment
phase. Essentially, the blocks from the search phase
are further refined using an efficient search algo-
rithm based on overlapping k-mers, the BG algo-
rithm (Salmela et al., 2006). The workflow of the
alignment phase is shown in the right section of the
Figure 1.
The BG algorithm is based on the BNDM algo-
rithm (Navarro and Raffinot, 2000) for a single pat-
tern. The idea is to construct a generalized pattern that
represents a group of patterns. For example, the group
of patterns, acgt, aacc, and gttt can be represented
by the generalized pattern: [a,g][a,c,t][c,g,t][c,t]. The
BG algorithm finds the generalized pattern represent-
ing overlapping k-mers. If k = 2 in the example
above, the corresponding generalized pattern is given
by [ac,aa,gt][cg,ac,tt][gt,cc,tt]. Each occurrence of
the generalized pattern is a candidate for a real match.
BG works as a filter and the candidate matches are
checked by an exact method. In practice, the BG al-
gorithm is very efficient (Salmela et al., 2006).
The overlappingk-mer patterns used by the BG al-
gorithm in our tool, which will be referred to as BG-
probes, are composed of 5 consecutive 7-mers taken
from the query sequences. The number of these BG-
probes taken from the query is chosen to be 0.6 times
the length of query sequence, and these BG-probes
are taken as evenly from the query as possible. This
BG-probe frequency has proven out to be suitable ac-
cording to our experiments.
The collection of database blocks resulting from
the initial search phase is scanned one block at a time
for occurrences of the BG-probes extracted from the
query sequence. This scanning uses the 7-mer en-
coded text.
When a BG-probe occurring in the query se-
quence is found from the database block, the positions
of the probe in the database block and query are saved
as a pair in a probe-hit list. After the scanning of the
database block is complete, the probe-hits are sorted
by difference values d
i
= pd
i
− pq
i
of each pair in as-
cending order. Here pd
i
is the position of i:th BG-
probe occurrence in the database block and pq
i
is the
position of the same BG-probe in the query sequence.
Following this sorting, the probe-hits are com-
bined in to what we call structured sets. If the d
i
of two consecutive probe-hits in the sorted list differ
by less than 10, these two probe-hits are put into the
same structured set. This maximum difference value
has worked well in our experiments.
After all the probe-hits have been assigned to a
structured set, the sets which have less than mt probe-
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
84
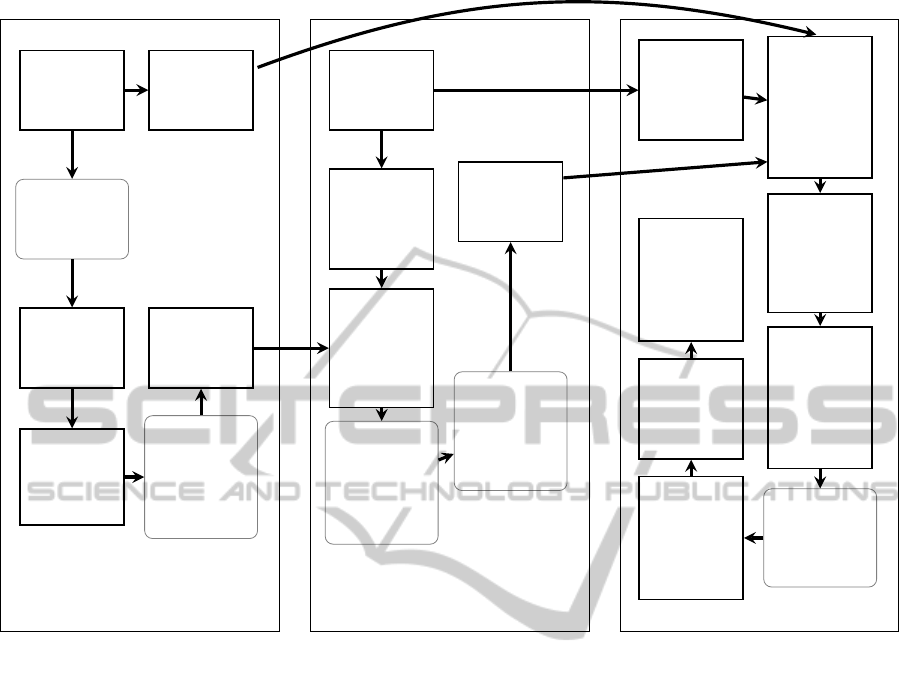
Database
sequences
Block size
Blocks
Collect
blockwise
AC-probe
locations
ACdisc:
discard
most
common
probes
Resulting
AC-index
structure
Using BG
algorithm,
search
BG-probes
within
blocks
k-mer
encoded
text
Query
sequences
Collect
AC-probes
from the
query
Sum
matching
AC-probes
in each
block
Threshold:
mp *
approved
AC-probes
in pattern
Threshold:
hits/block
≥ rp *
hits in
best block
Collection
of good
blocks
Obtain
BG-probes
from
pattern
Sort BG-
probes by
position
difference
value
Combine
related
probe-
hits to
structured
sets
Threshold:
# of pairs
in a set
≥ mt
Sort
probe-hits
in sets by
appearance
in query
Expand
hits, ap-
proximate
gaps
Report
sites, mis-
matches
+ gaps
as output
INDEX
INITIAL SEARCH ALIGNMENT
Figure 1: The workflow of the GAST tool. Left section describes the creation of block-addressing q-sample index, mid section
describes initial search phase, right section describes alignment phase. Parameters and thresholds in gray, rounded boxes.
hits are discarded, and the rest are sorted by the pq
i
value of each pair in ascending order. Resulting struc-
tured set correspondsto an aligned region between the
query sequence and the database block. The thresh-
old value mt can be specified by the user and it has an
effect on the minimum size of an alignment.
Lastly, the structured sets are expanded from both
ends as long as the nucleotides between the query
sequence and the database sequence match. If the
d
i
value of two consecutive probe-hits differ, corre-
sponding number of gaps are considered to exist be-
tween probe-hits. The placing of gaps resulting in
least mismatches is chosen.
Repeating these steps for all the structured sets,
blocks and queries, approximate alignments for each
of the queries are obtained. If desired, it is also pos-
sible to perform this search for the reverse comple-
ment of the given query sequence. As the output,
GAST reports the start and end sites of the alignment
in both the query and the database. The number of
mismatches and gaps is also reported. Additionally,
there is an option to output the actual approximated
alignment.
2.4 Parameters
There are five parameters used by GAST, which are
adjustable by the user. Four of these, the parameters
ACdisc, mp, rp, and mt act as threshold or cutoff val-
ues in different stages. The fifth is the size of a block
in the index structure.
The block size b directly affects the size of the
index, therefore affecting the level of memory con-
sumption. A suitable value for the block size in our
experiments has been 500, 000 nucleotides. Larger
blocks result in smaller relative differences between
the number of AC-probes found in them and cause
larger areas of text to be passed on to the alignment
phase. A smaller block size results in larger number
of blocks, resulting in larger index files. It is not ad-
vised to set the block size to be smaller than the length
of the query sequence searched, as the query may be
then splitted between more than two blocks.
The ACdisc parameter controls the number of dif-
ferent AC-probes considered to be approved, mean-
ing that they will be used to compare the query se-
quence with the blocks of the text. The default value
GAST, A GENOMIC ALIGNMENT SEARCH TOOL
85

for ACdisc is 0.1, resulting in the most common tenth
of the AC-probes to be discarded. Lower ACdisc val-
ues will cause more common AC-probes to be taken
into account. As result, the average AC-probe will
cause less distinction between blocks, which is not
desirable. With higher values, a smaller amount of
AC-probes will be taken into account, causing less
approved AC-probes to be found from query, which
makes the initial search phase less tolerant to differ-
ences between the query and the text. Suitable values
in our experiments have been in the range [0.001, 0.1].
The parameter mp defines the minimum amount
of approved AC-probes a block has to have in com-
mon with the query sequence in order to be con-
sidered potential. The default value for mp is 0.1.
Lower mp values allow blocks that differ more from
the query sequence to be taken into account. Higher
mp values leave less room for differences between
the approved AC-probes of the block and the query
sequence. Together the parameters ACdisc and mp
have a major effect on run times, especially when
very low values are chosen. Combination of ACdisc
values of 0.003 or smaller and mp values of 0.05 or
smaller should be avoided, as this leads to increase
of run times by 40–100 orders of magnitude. If the
user wishes to take initially less promising blocks in
to the final alignment stage, we suggest values ACdisc
= 0.01 and mp = 0.07 to be used. More such balanced
combinations are shown in Section 3.
As the final cutoff affecting the initial search
phase, each block has to contain at least a portion rp
of maximum amount of approved AC-probes match-
ing between a single block and the query sequence,
in order to be considered potential. The default value
for rp is 0.8. As the occurrences of approved AC-
probes can be scattered to distant, non-related regions
within a block, values very close to 1 may lead to
situations where blocks resulting in more more sat-
isfactory alignments with the query are discarded. In
our experiments, rising the rp value above 0.8 has de-
creased the run times only by 10–20%, and we do not
advise to use values higher than this.
The alignment phase has one adjustable parameter
value mt, which is the minimum amount of probe-hits
a structured set has to have for it to be considered to
correspond to an alignment between the query and the
text sequence. The default value for mt is 60. This
parameter effectively defines the minimum length of
a constructed alignment. If user wants to take into
account very short alignments between the query se-
quence and the database text block, smaller values can
be used. In our experience, changing the value of mt
from 60 to 10 causes run time increase by slightly less
than 10%. However, if shorter alignments are not ex-
plicitly desired, the default value is recommended, as
smaller values will result in larger amount of short,
probably less interesting alignments to be output.
Additionally, there are few built-in choices re-
garding values, which affect the function of our tool.
One of these is the length of the AC-probes, which
was chosen to be 10 nucleotides in addition to the
dinucleotide AC. If the length of the probe is in-
creased, this increases the size of the index and re-
quires database sequence to have longer identical re-
gions with the query sequence. Shorter AC-probes
will occur more commonly, causing less distinction
between blocks.
Similar balancing is required for the length and
amount of k-mers included in a single BG-probe,
which is used by the BG algorithm in the alignment
phase. The q must be small enough for the size of the
k-mer encoded database text files to be manageable.
However it is beneficial to have BG-probes, which
have a high probability of being unique in a block.
Our choices for the length of the AC-probes and BG-
probes are balanced compromises, which have proven
out to work well in our experiments.
3 RESULTS
Our algorithm was compared with the algorithms
Mega BLAST (Zhang et al., 2000) and BLAT (Kent,
2002). For GAST and Mega BLAST, searches were
made against a database consisting of the whole hu-
man genome received from the Ensembl genome
database (Hubbard, T. J. P. et al. , 2007). The release
in question was based on the NCBI 36 assembly of the
human genome. In the case of BLAT, the system used
for the runs lacked the memory to perform searches
against the whole human genome. Therefore, an-
other set of searches with BLAT, Mega BLAST, and
GAST were performed against the chromosome 1 of
the same genome. All the runs were performed on a
machine with 1GB DDRII SDRAM (667MHz) and an
Intel Core 2 Duo T5500 (1.66 GHz) processor, run-
ning Ubuntu 7.04. All the run times in this section
are times used by the program itself and any library
subroutines it calls. The tests were later repeated on
another machine with 6 GB of RAM in order to elim-
inate possible paging effects. No bias of this sort was
detected.
The AC-index described in the previous section
was created, using block size 500, 000, AC-probe
length 12 and ACdisc value 0.1. In addition, the
database files were encoded with 7-mers. These steps
were performed for the full genome and for the chro-
mosome 1 separately.
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
86

Table 1: The run times for the preparatory steps of algorithms BLAT, Mega BLAST and GAST and the sizes of the structures
created by these steps.
Preprocessing Database Time Size
GAST, index full genome 287.46 s 79.5 MB
GAST, encode full genome 351.87 s 5.7 GB
Mega BLAST, formatdb full genome 157.59 s 734.8 MB
BLAT, makeOoc full genome 108.57 s 165.7 kB
GAST, index chromosome 1 25.65 s 10.3 MB
GAST, encode chromosome 1 26.49 s 471.6 MB
Mega BLAST, formatdb chromosome 1 12.44 s 59.0 MB
BLAT, makeOoc chromosome 1 8.45 s 3.1 kB
Figure 2: The run times for algorithms BLAT, Mega
BLAST and GAST with variable pattern lengths. For each
length, the algorithms were run with 250 randomly selected
queries. Only the searches with patterns long enough to
produce reliable results were included in the comparison.
For Mega BLAST, the database was preprocessed
with the formatdb tool, using file type “nucleotide”,
parsing sequence IDs and creating indexes. Databases
including the full genome and the one including only
chromosome 1 were preprocessed separately. For
BLAT, an index structure was created by collecting
over occurring 11-mers from the chromosome 1, us-
ing the -makeOoc parameter with -repMatch=1024.
The run times for preprocessing and the sizes of the
created structures are listed in Table 1. BLAT is
fastest in the creation of its indexes and it also has the
smallest indexes of the three. GAST builds smaller
index structures than Mega BLAST, but takes longer
creating them. The k-mer encoding of the text, per-
formed by GAST, is the most time and space consum-
ing of all these preprocessing steps. It is noteworthy,
that all of these preprocessing steps are of such na-
ture that they have to be performed only once for each
genome or other collection of database text files.
Run times were studied as a function of the length
of a query and as a function of the number of queries
to be mapped. Searches studying the effect that the
length of query has on run time were executed with
lengths varying from 10 to 10000 nucleotides. For
each length, 250 exact query strings were randomly
picked from the first chromosome. The algorithms
BLAT, Mega BLAST, and GAST were used to search
these queries in the first chromosome. BLAT was
run using the created over occurring 11-mer file and
using fast DNA/DNA remapping, which causes the
search to not allow introns, improving the speed of
the search remarkably. Mega BLAST was run using
the query sequence length as the minimum score of a
hit to be reported, causing it to report exact matches
only. GAST was run using the default parameter val-
ues ACdisc = 0.1, mp = 0.1, rp = 0.8, and mt = 60.
The run times for the three algorithms searching the
patterns are shown in Figure 2. Mega BLAST found
queries of length of 40 and greater, BLAT, 80 or
greater, and GAST 160 or greater reliably enough to
produce reportable results. Searches with shorter pat-
terns than these were excluded from the comparison.
As can be seen in Figure 2, BLAT was the fastest
tool with very short patterns of length 160 and be-
low. With pattern lengths of 320 and above, GAST
was the fastest tool of the three. The run time de-
pendence on the number of queries to be mapped was
studied using lengths of 1000 and 5000 and number
of queries increasing from 1 to 5000. Sets of pat-
terns were chosen randomly from the whole genome
and from the first chromosome separately. GAST
and Mega BLAST were run using the patterns chosen
from the whole genome and first chromosome sep-
arately. Mega BLAST was run using parameters -s
1000 and -X 1, where -s is the minimum score to be
reported and -X is the X drop off value for gapped ex-
tension. GAST was run using the same parameters as
in the previous test. BLAT was run for the chromo-
some 1 sets, using the created over occurring 11-mer
file and fast DNA/DNA remapping. The results for
the whole genome are shown in Figure 3 and results
for the chromosome 1 are shown in Figure 4.
GAST, A GENOMIC ALIGNMENT SEARCH TOOL
87
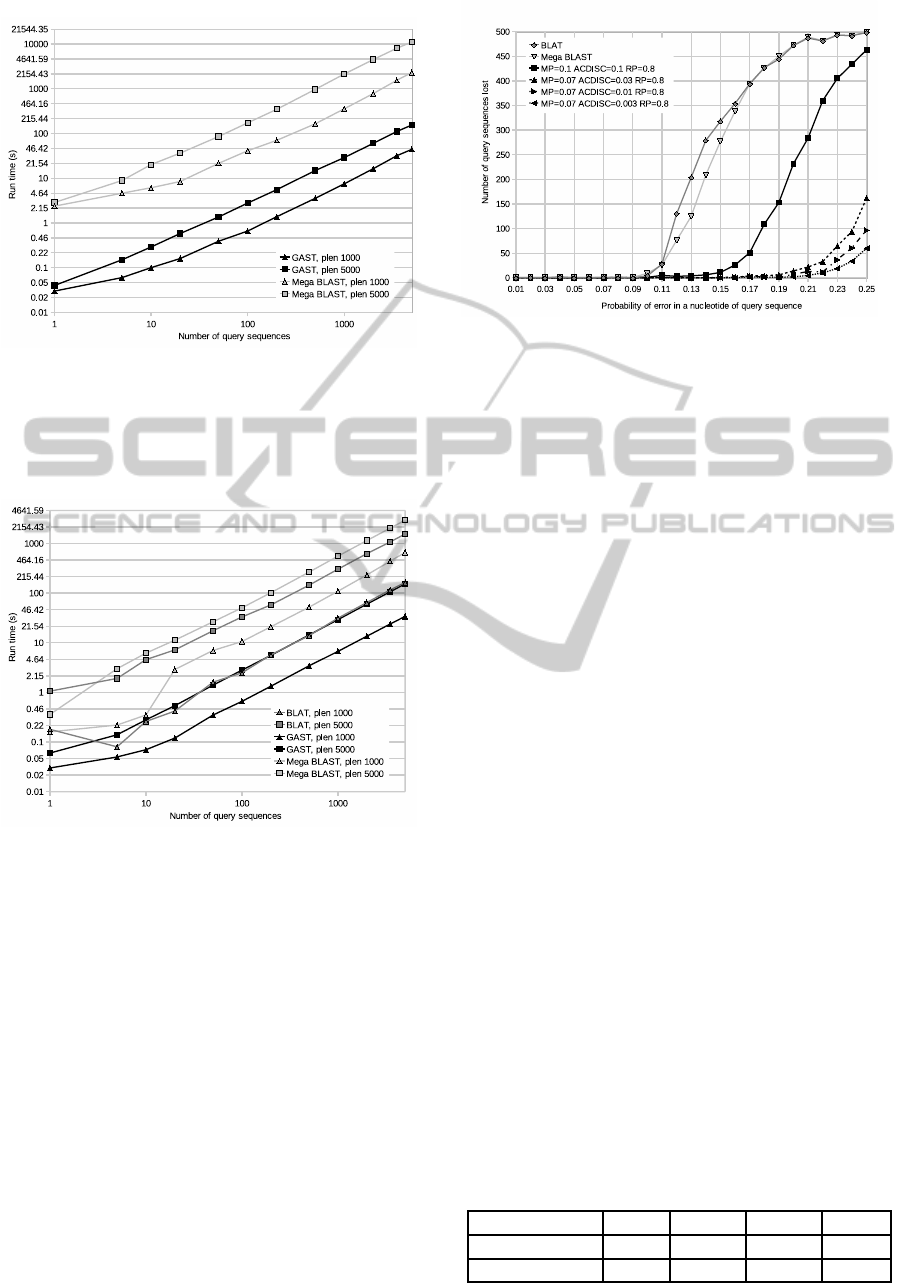
Figure 3: The run times for algorithms Mega BLAST and
GAST searching a variable number of queries from the
whole genome of Homo sapiens. For each number of pat-
terns, there were two sets to be searched, one consisting of
patterns of length 1000 and the other consisting of patterns
of length 5000.
Figure 4: The run times for algorithms BLAT, Mega
BLAST and GAST searching a variable number of patterns
from the chromosome 1 of Homo sapiens. For each number
of query sequences, there were two sets to be searched, one
consisting of patterns of length 1000 and the other consist-
ing of patterns of length 5000.
The run times shown in Figures 3 and 4 increase
linearly as the number of patterns increase, as sus-
pected. The difference caused by the change of
database size is very notable. Comparing the average
run times for query sequence length of 1000, GAST
was 50 times faster than Mega BLAST on the full
genome, but only 18.3 times faster on the chromo-
some 1. For query sequence length of 5000, the cor-
responding numbers are 72.1 and 19.1.
The error tolerances of BLAT and Mega BLAST
were compared to the error tolerances of GAST, us-
ing various parameter values. 25 sets of 500 query se-
quences of length 5000 were randomly selected from
Figure 5: The amount of patterns BLAT, Mega BLAST and
GAST did not find when searching with pattern sets with
error probabilities ranging from 0.01 to 0.25. For GAST,
the default parameters and three additional parameter sets
with mp = 0.07 and ACdisc = [0.01, 0.03, 0.003] were used.
the first chromosome. Each of these sets had a fixed
error probability, ranging from 0.01 to 0.25, where
an error probability e means that each nucleotide in a
pattern has a probability e to to be randomly mutated.
Each mutation had a probability 0.5 to be a substitu-
tion, 0.25 to be an insertion and 0.25 to be a deletion.
Each algorithm was run with all the sets sepa-
rately. Parameter values for BLAT were the same as
in the previous tests. Mega BLAST was run with the
default -s parameter value of 0, to get reports from
all hits. The parameter sets used with GAST, and the
number of patterns that were not found in the runs
can be seen in Figure 5. Pattern was considered to
be found if the algorithm reported a hit within the se-
quence length from the original pattern site.
As can be seen in Figure 5, Mega BLAST and
BLAT both started quickly losing approximate occur-
rences of patterns with error rate of 0.12 and above.
GAST started losing approximate occurrences of pat-
terns at the same rate with a higher error rate of 0.17
and above, when using default parameters. As the fig-
ure shows, it is possible to further increase the error
tolerance of GAST by selecting smaller mp value of
0.07 and adjusting the ACdisc value gradually. The
run times are still relatively low with ACdisc values
0.03 and 0.01, as can be seen in the Table 2. As
ACdisc was further decreased to a value of 0.003, a
notable increase in run times occurred.
Table 2: Average GAST run times depending on parame-
ters. Sets of 500 patterns of length 5000 with error proba-
bilities varying from 0.01 to 0.25. The rp parameter values
were 0.8 for all sets.
mp 0.1 0.07 0.07 0.07
ACdisc 0.1 0.03 0.01 0.003
Avg. run time 7.94s 10.82s 15.33s 46.5s
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
88
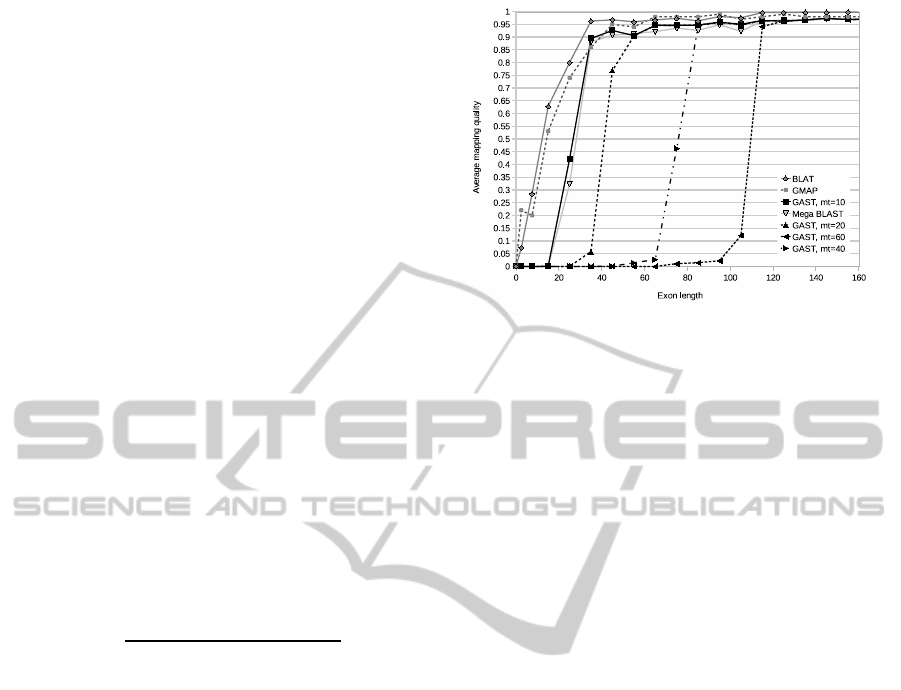
Lastly, the run times and exon mapping quality
were studied for a set of 6721 cDNA sequences, cor-
responding to various transcripts originating from hu-
man chromosome 1. The sequences were retrieved
from the BioMart database (OICR and EBI, 2010) and
were 2000 nucleotides long on average. The starting
and ending positions of exons in the sequences were
also retrieved. All the three tools and the GMAP tool
were run to map these sequences to the human chro-
mosome 1.
Mega BLAST was run using an index created as
described before. The parameters were default apart
from using this index structure. GAST was run with
default parameters apart from the match threshold pa-
rameter mt, which was given values 60, 40, 20 and
10 in subsequent runs. BLAT was run with the same
parameters as before, but the fast DNA/DNA remap-
ping, which does not allow introns, was disabled for
this task. GMAP was run with -B paramater value of
2, meaning that both the text and the index made of it
were read to memory, and with parameter -A to cal-
culate alignment between the query and the database.
The run times for these runs are in the Table 3.
A quality measure Q
exon
for the exon mappings
produced was calculated for all the resulting map-
pings as follows:
Q
exon
=
L
e
− (|m
s
− e
s
| + |m
e
− e
e
|)
L
e
(1)
where L
e
is the exon length, m
s
and m
e
are the start
and end positions of the mapping, and e
s
and e
e
are
the start and end positions of the exon, respectively.
The score obtained from the mapping giving the high-
est score, out of the mappings of the cDNA in ques-
tion, is saved for each exon. For each run, averages of
scores Q
exon
were calculated for exon length ranges
of 0–5, 5–10, 10–20, 20–30, ..., 150–160. These av-
erages are in Figure 6.
The run times given in Table 3 show very remark-
able differences between the four tools, GAST being
the fastest. The notable increase of run time for BLAT
is most likely caused by disabling fast DNA/DNA
remapping. The quality of exon mappings produced
by BLAT and GMAP were highest out of all tools.
Mappingsproduced by GAST with mt = 10 and Mega
BLAST gained slightly lower quality scores, espe-
cially when mapping very short exons. With higher
mt values, GAST was not able to produce mappings
for shorter exons.
Figure 6: Average exon mapping quality as the function of
exon length for the tools GAST, BLAT, Mega BLAST, and
GMAP. The performance of GAST is shown with different
match threshold parameter values.
4 CONCLUDING REMARKS
According to our results, GAST is an efficient and ac-
curate search tool for common genomic search prob-
lems. Concerning the speed, our algorithm performs
best with long sequences, large databases, and large
query sets. As the length of either the database or
the length of the pattern increases, the magnitudes of
differences between GAST, Mega BLAST, and BLAT
increases.
The drawbacks of such increases in efficiency are
the space and time requirements of the preprocessing
steps, namely the creation of the AC-index and the k-
mer encoding of the database files. However, as the
preprocessing steps are required to be done only once
for each genome, or other set of sequence files, the
authors see this as a relatively small problem. De-
pending on the sizes of the patterns and the database
files, GAST would pay back the computational re-
quirements of the preprocessing steps after a few hun-
dred search queries.
The usage of the AC-index causes requirements
for the lengths of the queries. According to our stud-
ies, it is best to use GAST for patterns of at least 200–
300 nts in length. However, in a long enough tran-
script or cDNA, the exons can be shorter than this, de-
pending on the chosen mt parameter. The mentioned
nucleotide requirement is just to make sure that there
are enough AC probes in the sequence for it to be ini-
tially mapped to the correct block.
With longer sequences of length 5000, GAST
has proven out to be more error tolerant than Mega
BLAST and BLAT, as can be seen in Figure 5. It is
also possible to further improve the error tolerance of
our search tool by tweaking the parameters ACdisc
GAST, A GENOMIC ALIGNMENT SEARCH TOOL
89

Table 3: The run times for the mapping of 6721 cDNA sequences on human chromosome 1, allowing introns. GASTXX
stands for a GAST run with XX for the parameter mt.
BLAT Mega BLAST GMAP GAST10 GAST20 GAST40 GAST60
286m 40.3s 45m 19.2s 14m 52.4s 1m 13.7s 1m 10.6s 1m 8.4s 1m 8.3s
and mp, while still retaining the speed advantage over
BLAT and Mega BLAST.
Additionally, the quality of the exon mapping pro-
duced by GAST is on par with that of Mega BLAST
and comparable to the mappings done by BLAT and
GMAP. However, the mapping was multiple orders of
magnitude faster with GAST.
In the future, we would like to compare more
genomic alignment and mapping tools with GAST.
The possibility of using statistical methods to derive
more optimal parameter values could also be exam-
ined. Studies with different dinucleotides and trinu-
cleotides forming the probes for the index could also
be done. Lastly, the compatibility of the output data
formats should be developed further on to allow eas-
ier pipelining of GAST with other methods, possibly
further refining the areas initially suggested by GAST.
ACKNOWLEDGEMENTS
We thank Sami Khuri and anonymous referees for
comments that helped us to improve the presenta-
tion of this paper. The work was supported by the
Academy of Finland (grant 134287).
REFERENCES
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and
Lipman, D. J. (1990). Basic local alignment search
tool. Journal of Molecular Biology, 215(3):403–410.
Harper, C. A., Huang, C. C., Stryke, D., Kawamoto, M.,
Ferrin, T. E., and Babbitt, P. C. (2006). Comparison
of methods for genomic localization of gene trap se-
quences. BMC Genomics, 7:236.
Hubbard, T. J. P. et al. (2007). Ensembl 2007. Nucleid Acid
Res., 35:D610–D617.
Karlin, S. and Burge, C. (1995). Dinucleotide relative abun-
dance extremes: a genomic signature. Trends in Ge-
netics, 11(7):283–290.
Kent, W. J. (2002). BLAT - The BLAST-like alignment tool.
Genome Res., 12:656–664.
Manber, U. and Wu, S. (1994). GLIMPSE: A tool to
search through entire file systems. Proceedings of the
USENIX Winter Conference, pages 23–32.
Navarro, G. and Raffinot, M. (2000). Fast and flexible string
matching by combining bit-parallelism and suffix au-
tomata. ACM Journal of Experimental Algorithms 5,
4:1–36.
NCBI (2009). www.ncbi.nlm.nih.gov/BLAST/ (cited Mar
24, 2009), BLAST: Basic Local Alignment Search
Tool (on-line).
Ning, Z., Cox, A. J., and Mullikin, J. C. (2001). SSAHA:
A Fast Search Method for Large DNA Databases.
Genome Res., 11:1725–1729.
OICR and EBI (2010). www.biomart.org (cited May 3,
2010), BioMart Project (on-line).
Salmela, L., Tarhio, J., and Kyt¨ojoki, J. (2006). Multi-
pattern string matching with q-grams. ACM Journal
of Experimental Algorithms, 11(1).
Sutinen, E. and Tarhio, J. (1996). Filtration with q-samples
in approximate string matching. In Proceedings of
the 7th Annual Symposium on Combinatorial Pattern
Matching (CPM ’96). Lecture Notes in Computer Sci-
ence, 1075:50–63.
Wu, T. D. and Watanabe, C. K. (2005). GMAP: a genomic
mapping and alignment program for mRNA and EST
sequences. Bioinformatics, 21(9):1859–1875.
Zhang, S.-H. and Yang, J.-H. (2005). Conservation versus
variation of dinucleotide frequencies across genomes:
Evolutionary implications. Genome Biology, 6, P12.
Zhang, S.-H. and Yang, J.-H. (2008). Characteris-
tics of oligonucleotide frequencies across genomes:
Conservation versus variation, strand symmetry,
and evolutionary implications. Nature Precedings,
hdl:10101/npre.2008.2146.1.
Zhang, Z., Schwartz, S., Wagner, L., and Miller, W. (2000).
A greedy algorithm for aligning DNA sequences.
Journal of Computational Biology, 7:203–214.
BIOINFORMATICS 2011 - International Conference on Bioinformatics Models, Methods and Algorithms
90
