
PERFORMANCE STUDY OF PARALLEL HYBRID MULTIPLE
PATTERN MATCHING ALGORITHMS FOR BIOLOGICAL
SEQUENCES
Charalampos S. Kouzinopoulos
1
, Panagiotis D. Michailidis
2
and Konstantinos G. Margaritis
1
1
University of Macedonia, Thessaloniki, Macedonia, Greece
2
University of Western Macedonia, Kozani, Florina, Greece
Keywords:
Algorithms, Multiple pattern matching, Parallel computing, Hybrid, OpenMP, MPI, Biological sequence
databases.
Abstract:
Multiple pattern matching is widely used in computational biology to locate any number of nucleotides in
genome databases. Processing data of this size often requires more computing power than a sequential com-
puter can provide. A viable and cost-effective solution that can offer the power required by computationally
intensive applications at low cost is to share computational tasks among the processing nodes of a high per-
formance hybrid distributed and shared memory platform that consists of cluster workstations and multi-core
processors. This paper presents experimental results and a theoretical performance model of the hybrid im-
plementations of the Commentz-Walter, Wu-Manber, Set Backward Oracle Matching and the Salmela-Tarhio-
Kyt¨ojoki family of multiple pattern matching algorithms when executed in parallel on biological sequence
databases.
1 INTRODUCTION
Multiple pattern matching of nucleotides and amino
acid sequence patterns in biological sequence
databases is an important application in bioinformat-
ics that can identify diagnostic patterns or motif to
characterize protein families. It can also detect or
demonstrate homology between new sequences and
existing families of sequences. This way, it helps to
predict the secondary and tertiary structures of new
sequences which is an essential prelude to molecular
evolutionary analysis (Chaichoompu et al., 2006).
Given a sequence database or input string T =
t
1
t
2
. . . t
n
of length n and a finite set of r patterns
P = p
1
, p
2
, . . . , p
r
, where each p
i
is a string p
i
=
p
i
1
p
i
2
. . . p
i
m
of length m over a finite character set Σ
and the total size of all patterns is denoted as |P
r
|, the
task is to find all occurrences of the patterns in the
sequence database.
For the experiments of this paper, the simple,
efficient and widely used Commentz-Walter (CW)
(Commentz-Walter, 1979), Wu-Manber (WM) (Wu
and Manber, 1994), Set Backward Oracle Match-
ing (SBOM) (Navarro and Raffinot, 2002) and
the Salmela-Tarhio-Kyt¨ojoki family (Salmela et al.,
2006) of multiple pattern matching algorithms were
used. The Commentz-Walter algorithm is substan-
tially faster in practice than the Aho-Corasick (Aho
and Corasick, 1975) algorithm, particularly when
long keywords are involved (Watson, 1995)(Wu and
Manber, 1994). Wu-Manber is considered to be a
practical, simple and efficient algorithm for multiple
keyword matching (Navarro and Raffinot, 2002). The
Set Backward Oracle Matching algorithm appears to
be very efficient when used on large keyword sets. It
has the same worst case complexity as Set Backward
Dawg Matching but uses a much simpler automaton
and is faster in all cases (Navarro and Raffinot, 2002).
Finally, Salmela-Tarhio-Kyt¨ojoki is a recently intro-
duced family of algorithms that has a reportedly good
performance on specific types of data (Kouzinopoulos
and Margaritis, 2010). For further details on the above
algorithms the reader is referred to (Kouzinopoulos
et al., 2011) and the original references.
Over the last decades, as the amount of biolog-
ical sequence data available in databases worldwide
grows at an exponential rate, researchers continue to
require faster and more powerful search algorithms.
The sequential computer programs can not deal effi-
ciently with both execution time and memory require-
ments for large-scale multiple pattern matching algo-
rithms. With these two constraints in mind, a new
182
S. Kouzinopoulos C., D. Michailidis P. and G. Margaritis K..
PERFORMANCE STUDY OF PARALLEL HYBRID MULTIPLE PATTERN MATCHING ALGORITHMS FOR BIOLOGICAL SEQUENCES.
DOI: 10.5220/0003769801820187
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2012), pages 182-187
ISBN: 978-989-8425-90-4
Copyright
c
2012 SCITEPRESS (Science and Technology Publications, Lda.)

class of high performance computing platforms and
tools appeared in the last few years, including clus-
ters of workstations and multicore processors, in or-
der to reduce the high time and memory requirements
on large-scale biological databases.
Parallel implementations and experiments for the
cases of pairwise sequence alignment and multiple
sequence alignment have been presented in the re-
search literature for distributed memory platforms
(Li, 2003), (Li and Chen, 2005), (Boukerche et al.,
2007), (Jacob et al., 2007) and for shared mem-
ory platforms (Cuvillo et al., 2003), (Chaichoompu
et al., 2006), (Rashid et al., 2007), (Zomaya, 2006).
The above implementations are based on the paral-
lelization of known biological sequence analysis al-
gorithms such as the Smith-Waterman, Needleman-
Wunsch and ClustalW algorithms.
This paper presents hybrid implementations of
the Commentz-Walter, Wu-Manber, and the Salmela-
Tarhio-Kyt¨ojoki family of multiple pattern matching
algorithms on a hybrid distributed and shared mem-
ory architecture when executed on large biological
sequence databases. This technique could poten-
tially have a better performance than the traditional
distributed and shared memory parallelization tech-
niques. The current work differs from previous re-
search works in the fact that the proposed parallel
implementations for multiple sequence comparison
are based on multiple pattern matching algorithms
instead of biological sequence alignment algorithms.
The goal of this paper is to investigate the efficiency
of the hybrid parallel implementations of multiple
pattern matching algorithms on large biological se-
quence databases in a systematic and unified way.
2 PARALLEL
IMPLEMENTATION
The presented hybrid parallel implementation of the
algorithms combines the advantages of both shared
and distributed memory parallelization on a cluster
system consisting of multiple interconnected multi-
core computers using a hierarchical model. At the
first level, parallelism is implemented on the multi-
core computers using MPI where each MPI process is
assigned to a differentnode. In the next level, the MPI
processes spread parallelism to the local processors
by using a combined parallel work-sharing construct
for each computation, namely a parallel for directive;
each OpenMP thread is assigned to a different proces-
sor core. Figure 1 presents a pseudocode of the hybrid
implementation of the algorithms.
The following assumptions were made for the dis-
tributed memory parallelization: To enable communi-
cation between the cluster nodes, the master-worker
model was used, as it is very appropriate for pat-
tern matching. With this model, the master process
creates a series of separate but identical worker pro-
cesses which perform any sequential multiple pattern
matching algorithm on local data concurrently using
synchronous communication operations of the MPI li-
brary.
Main procedure
main()
{
1. Initialize MPI and OpenMP routines;
2. If (process==master) then call master(); else call
worker();
3. Exit MPI operations;
}
Master sub-procedure
master()
{
1. Send the text offset and the block size to each of
the workers;
2. Receive the results (i.e. matches) from all workers;
3. Print the total results;
}
Worker sub-procedure
worker()
{
1. Preprocess the pattern set;
2. Receive the offset of the text and the block size;
3. Open the sequence database from the local disk and
store the local subtext (from offset to offset +
block size) in memory;
4. Call the chosen multiple pattern matching algorithm
passing a pointer to the subtext in memory;
5. Divide the subtext among the available threads
6. Determine the number of matches from each thread
7. Send the results (i.e. matches) to master
}
Figure 1: Pseudocode of the hybrid implementation.
The complete biological databases and patterns
are distributed to each process using the NFS protocol
and are preloaded to memory before the preprocess-
ing phase of the algorithms begins, while the master
maintains a text offset that indicates the current po-
sition of the sequence database. Due to the homo-
geneity of the cluster nodes and the balanced data set
to be processed, a static distribution of the text offset
among the workers was chosen. Let N be the num-
ber of the available worker nodes, then the sequence
database will be decomposed in to ⌈
n
N
⌉ + N(m − 1)
successive characters prior to the execution of the al-
gorithms. The additional N(m− 1) pattern characters
between successive parts of the database ensure that
PERFORMANCE STUDY OF PARALLEL HYBRID MULTIPLE PATTERN MATCHING ALGORITHMS FOR
BIOLOGICAL SEQUENCES
183

each process has all the required data.
As opposed to distributed memory parallelization,
shared memory parallelization does not actually in-
volve a distribution of data since the entire data set is
stored in a common memory area where it can be ac-
cessed by all processing cores. To control the way the
iterations of the parallel implementations of the algo-
rithms are assigned to threads, OpenMP provides the
static, dynamic and guided scheduling clauses. Since
the pattern locations were generally balanced across
the data set and the use of dynamic scheduling usu-
ally incurs high overheads and tends to degrade data
locality (Ayguad´e et al., 2003), the static scheduling
clause of OpenMP was used as it was expected to be
best suited for the experiments of this paper. A similar
conclusion was drawn in (Kouzinopoulos and Mar-
garitis, 2009) where the static scheduling clause with
the default chunk size had a better performance than
the dynamic and guided scheduling clauses for two
dimensional pattern matching algorithms.
In many scenarios it is advantageous to estimate
the performance of a parallel system in order to de-
termine the efficiency of the implementation and to
verify the experimental results. The total execution
time of a parallel implementation of a multiple pat-
tern matching algorithm is equal to the I/O time to
read the data set from the file system, the preprocess-
ing time, the searching time and the communication
time. The I/O is performed offline before the exe-
cution of the algorithms and therefore its time is not
included. The preprocessing phase of most multiple
keyword matching algorithms is complex in nature
and cannot be efficiently distributed among different
processor cores, it is therefore performed sequentially
by each worker node. Finally, the search phase of the
algorithms is executed in parallel by t threads on each
of the N nodes of the computer cluster:
T
par
= T
preprocess
+ T
comm
+
T
search
N ×t
(1)
The communication time α of the distributed
memory implementation of the multiple pattern
matching algorithms is the summation of two com-
ponents: latency and transmission time. Latency is a
fixed startup overhead time needed to prepare send-
ing a message from one node to another. The time to
actually transmit a message is also fixed. The total
communication time T
comm
to transmit 2 messages for
the N nodes of the cluster is defined as:
T
comm
= 2 × N × α (2)
The search time consists of the actual time re-
quired by the cluster nodes to locate the patterns in
the sequence database in parallel and an overhead β
introduced by the OpenMP parallel, for and reduc-
tion constructs. The parallel computation time T
par
of the hybrid implementation of the multiple pattern
matching algorithms will then be equal to:
T
par
= T
preprocess
+ 2× N × α+
T
search
N × t
+ β (3)
To find the value of α, a simple program was used
that continuously transmitted a number of messages
between two nodes using MPI. Based on this test, and
for the specific experimental setup, α was equal to
0.00011 seconds. The measured value of α also in-
cludes the overhead introduced by the MPI Send and
MPI Recv functions of MPI. The value of β was es-
timated for the reference hardware as being equal to
0.00015 seconds.
3 EXPERIMENTAL RESULTS
The data set used consisted of the genome of Es-
cherichia coli from the Large Canterbury Corpus with
a size of n = 4.638.690 and an alphabet size Σ = 4, the
SWISS-PROT Amino Acid sequence database with a
size of n = 182.116.687 characters and an alphabet
size Σ = 20, the FASTA Amino Acid (FAA) of the A-
thaliana genome with a size of n = 11.273.437 char-
acters and an alphabet size Σ = 20 and the FASTA
Nucleidic Acid (FNA) sequences of the A-thaliana
genome with a size of n = 118.100.062characters and
an alphabet size Σ = 4. The pattern set used consisted
of 10.000 patterns where each pattern had a length of
m = 8 characters.
Table 1: Preprocessing and total running time of the algo-
rithms for the E.coli and SWISS-PROTsequence databases.
E.coli SWISS-PROT
Algorithm Prepr. Running Prepr. Running
CW 0.014 2.868 0.044 18.378
WM 0.002 3.388 0.010 9.629
HG 0.062 1.987 0.137 4.869
SOG 0.059 1.999 0.044 5.969
BG 0.069 1.982 0.141 4.912
The experiments were executed on a homoge-
neous computer cluster consisting of 10 nodes with an
Intel Core i3 CPU with a 2.93GHz clock rate and 4 Gb
of memory, a shared 4MB L3 cache and two micro-
processors cores, each with 64 KB L1 cache and 256
KB L2 cache. The nodes were connected using Re-
altek Gigabit Ethernet controllers. The Ubuntu Linux
operating system was used on all systems and dur-
ing the experiments only the typical background pro-
cesses ran. To decrease random variation, the time re-
sults were averages of 100 runs. All algorithms were
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
184
2
4
6
8
10
12
14
16
18
20
1 2 3 4 5 6 7 8 9 10
Speedup
Number of processing nodes (E.coli)
CW
WM
HG
SOG
BG
2
4
6
8
10
12
14
16
18
20
1 2 3 4 5 6 7 8 9 10
Speedup
Number of processing nodes (SWISS-PROT)
CW
WM
HG
SOG
BG
2
4
6
8
10
12
14
16
18
20
1 2 3 4 5 6 7 8 9 10
Speedup
Number of processing nodes (FAA)
CW
WM
HG
SOG
BG
2
4
6
8
10
12
14
16
18
20
1 2 3 4 5 6 7 8 9 10
Speedup
Number of processing nodes (FNA)
CW
WM
HG
SOG
BG
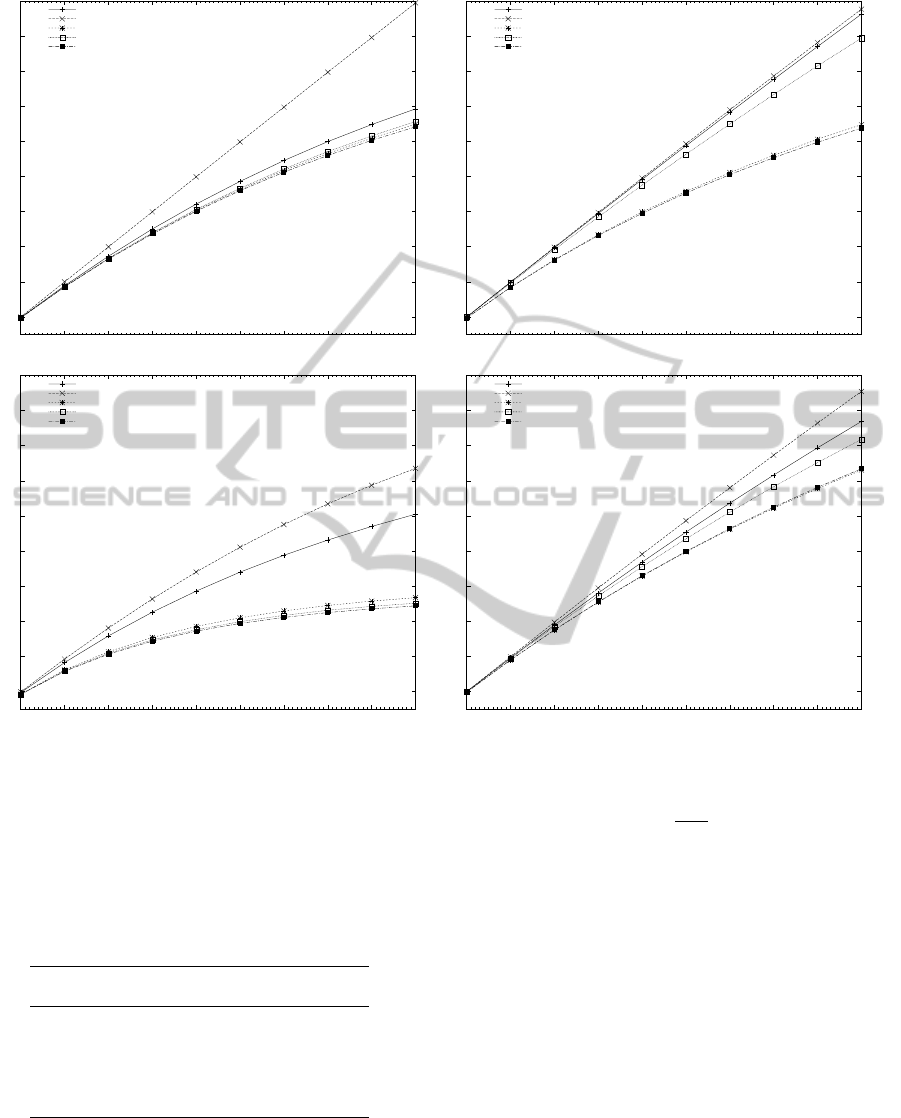
Figure 2: Estimated speedup of all algorithms with hybrid OpenMP/MPI for different number of processors with two cores.
implemented using the ANSI C programming lan-
guage and were compiled using the GCC 4.4.3 com-
piler with the “-O2” and “-funroll-loops” optimiza-
tion flags.
Table 2: Preprocessing and total running time of the algo-
rithms for the FAA and FNA sequence databases.
FAA FNA
Algorithm Prepr. Running Prepr. Running
CW 0.020 0.672 0.042 5.234
WM 0.013 0.476 0.007 4.939
HG 0.021 0.230 0.042 2.441
SOG 0.032 0.279 0.045 2.714
BG 0.024 0.232 0.040 2.437
Speedup S
p
refers to the running time of a parallel
algorithm T
par
over a corresponding execution time of
a sequential algorithm T
seq
when executed on a paral-
lel system with p processing elements.
S
p
=
T
seq
T
par
(4)
Based on the observations of the previous section
and the preprocessing and searching time of the se-
quential implementation of the algorithms as depicted
in Tables 1 and 2, the parallel speedup of the algo-
rithms can be estimated with a relatively good degree
of success, as shown in Figure 2. When comparing
Figures 2 and 3 it can be seen that the estimated values
describe the general tendency of the speedup of the
parallel implementations, but they were not as accu-
rate to predict the exact parallel speedup achieved by
most algorithms. To improve the accuracy of the cal-
culations, a more advanced performance prediction
model could be used. A number of factors exist that
can explain the difference between the estimated and
the experimental results. The time to access the mem-
ory by the OpenMP threads, the execution cost of in-
struction cache misses, coherence cache misses and
PERFORMANCE STUDY OF PARALLEL HYBRID MULTIPLE PATTERN MATCHING ALGORITHMS FOR
BIOLOGICAL SEQUENCES
185

2
4
6
8
10
12
14
16
18
20
1 2 3 4 5 6 7 8 9 10
Speedup
Number of processors (E.coli)
CW
WM
HG
SOG
BG
2
4
6
8
10
12
14
16
18
20
1 2 3 4 5 6 7 8 9 10
Speedup
Number of processors (SWISS PROT)
CW
WM
HG
SOG
BG
2
4
6
8
10
12
14
16
18
20
1 2 3 4 5 6 7 8 9 10
Speedup
Number of processors (FAA)
CW
WM
HG
SOG
BG
2
4
6
8
10
12
14
16
18
20
1 2 3 4 5 6 7 8 9 10
Speedup
Number of processors (FNA)
CW
WM
HG
SOG
BG
Figure 3: Measured speedup of all algorithms with hybrid OpenMP/MPI for different number of processors with two cores.
bus contentions (Liao and Chapman, 2007) as well
as optimizations of modern compilers can affect the
performance of a parallel hybrid implementation both
positively and negatively.
Figure 3 illustrates the performance increase of
the Commentz-Walter, Wu-Manber and the Salmela-
Tarhio-Kyt¨ojoki family of multiple pattern matching
algorithms using the proposed hybrid OpenMP/MPI
technique on a homogeneous cluster of 10 nodes with
a Core I3 processor on each node using 2 OpenMP
threads per node. As can be seen in the Figures, the
type of sequence database that is used can greatly af-
fect the performance of the parallel implementation
of the algorithms.
More specifically, for the E.coli sequence
database, the parallelization rate of the algorithms
increased linear in the number of cluster nodes.
The Wu-Manber algorithm was up to 19.2 times
faster than its sequential implementation while the
Commentz-Walter, HG, SOG and BG algorithms had
on average a 14.5 times better performance. The
speedup of the multiple pattern matching algorithms
was similar for the SWISS-PROT and the FNA se-
quence databases; the speedup of the SOG algo-
rithm was 15.3 and 13.5 respectively, of the HG,
BG and Wu-Manber algorithms was 12 on average
while the parallelization rate of the Commentz-Walter
algorithm was 10.7 and 6.4. Finally for the FAA
genome, the Wu-Manber and the Salmela-Tarhio-
Kyt¨ojoki family of multiple pattern matching algo-
rithms had a similar speedup of 8.4 on average while
Commentz-Walter had a parallelization rate of 3.7.
4 CONCLUSIONS
This chapter presented implementations and experi-
mental results of the Commentz-Walter, Wu-Manber
and the Salmela-Tarhio-Kyt¨ojoki family of multiple
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
186

pattern matching algorithms when executed in par-
allel on a hybrid distributed and shared memory ar-
chitecture. The algorithms were used to locate all
the appearances of any pattern from a finite pattern
set on four biological databases; the genome of Es-
cherichia coli from the Large Canterbury Corpus, the
SWISS-PROT Amino Acid sequence database and
the FASTA Amino Acid (FAA) and FASTA Nucleidic
Acid (FNA)sequences of the A-thaliana genome. The
pattern set used consisted of 100.000 patterns where
each pattern had a length of m = 8 characters.
It was concluded that the parallelization rate of
most multiple pattern matching algorithms depends
on the type of sequence database used. The par-
allel implementation of the algorithms had the best
speedup when used on the E.coli and the worst on
the FAA sequence database. It was also shown that
the Wu-Manber algorithm was up to 19.2 times faster
than its sequential implementation, the Commentz-
Walter was up to 14.5 times faster while the Salmela-
Tarhio-Kyt¨ojoki family of multiple pattern matching
algorithms had a speedup of up to 15.3 times.
The work presented in this chapter could be ex-
tended with a more accurate performance prediction
model as well as with experiments that use additional
parameters like patterns of varying length and larger
pattern sets. Since biological databases and sets of
multiple patterns are usually inherently parallel in na-
ture, future research could focus on the performance
evaluation of the presented algorithms when parallel
processed on modern parallel architectures such as
Graphics Processor Units.
REFERENCES
Aho, A. and Corasick, M. (1975). Efficient string matching:
an aid to bibliographic search. Communications of the
ACM, 18(6):333–340.
Ayguad´e, E., Blainey, B., Duran, A., Labarta, J., Mart´ınez,
F., Martorell, X., and Silvera, R. (2003). Is the sched-
ule clause really necessary in openmp? In Interna-
tional workshop on OpenMP applications and tools,
volume 2716, pages 147–159.
Boukerche, A., de Melo, A. C. M. A., Ayala-Rinc´on, M.,
and Walter, M. E. M. T. (2007). Parallel strategies for
the local biological sequence alignment in a cluster of
workstations. J. Parallel Distrib. Comput., 67:170–
185.
Chaichoompu, K., Kittitornkun, S., and Tongsima, S.
(2006). MT-clustalW: multithreading multiple se-
quence alignment. In IPDPS.
Commentz-Walter, B. (1979). A string matching algorithm
fast on the average. Proceedings of the 6th Collo-
quium, on Automata, Languages and Programming,
pages 118–132.
Cuvillo, J., Tian, X., Gao, G., and Girkar, M. (2003). Per-
formance study of a whole genome comparison tool
on a hyper-threading multiprocessor. In ISHPC, pages
450–457.
Jacob, A. C., Sanyal, S., Paprzycki, M., Arora, R., and
Ganzha, M. (2007). Whole genome comparison on a
network of workstations. In ISPDC’07, pages 31–36.
Kouzinopoulos, C. and Margaritis, K. (2009). Parallel im-
plementation of exact two dimensional pattern match-
ing algorithms using MPI and OpenMP. In 9th Hel-
lenic European Research on Computer Mathematics
and its Applications Conference.
Kouzinopoulos, C. and Margaritis, K. (2010). Experimental
Results on Algorithms for Multiple Keyword Match-
ing. In IADIS International Conference on Informat-
ics, pages 274–277.
Kouzinopoulos, C., Michailidis, P., and Margaritis, K.
(2011). Parallel Processing of Multiple Pat-
tern Matching Algorithms for Biological Sequences:
Methods and Performance Results. InTech.
Li, K.-B. (2003). ClustalW-MPI: ClustalW analysis using
distributed and parallel computing. Bioinformatics,
19(12):1585–1586.
Li, Y. and Chen, C.-K. (2005). Parallelization of multiple
genome alignment. In HPCC’05, pages 910–915.
Liao, C. and Chapman, B. (2007). Invited paper: A
compile-time cost model for openmp. In Proceedings
of the 21st International Parallel and Distributed Pro-
cessing Symposium.
Navarro, G. and Raffinot, M. (2002). Flexible pattern
matching in strings: practical on-line search algo-
rithms for texts and biological sequences. Cambridge
University Press.
Rashid, N. A., Abdullah, R., and Talib, A. Z. H. (2007). Par-
allel homologous search with hirschberg algorithm: a
hybrid mpi-pthreads solution. In Proceedings of the
11th WSEAS International Conference on Comput-
ers, pages 228–233, Stevens Point, Wisconsin, USA.
World Scientific and Engineering Academy and Soci-
ety (WSEAS).
Salmela, L., Tarhio, J., and Kyt¨ojoki, J. (2006). Multipattern
string matching with q -grams. Journal of Experimen-
tal Algorithmics, 11:1–19.
Watson, B. (1995). Taxonomies and toolkits of regular lan-
guage algorithms. PhD thesis, Eindhoven University
of Technology.
Wu, S. and Manber, U. (1994). A fast algorithm for multi-
pattern searching. pages 1–11. Technical report TR-
94-17.
Zomaya, A. (2006). Parallel Computing for Bioinformatics
and Computational Biology: Models, Enabling Tech-
nologies, and Case Studies. Wiley.
PERFORMANCE STUDY OF PARALLEL HYBRID MULTIPLE PATTERN MATCHING ALGORITHMS FOR
BIOLOGICAL SEQUENCES
187
