
MASon: MILLION ALIGNMENTS IN SECONDS
A Platform Independent Pairwise Sequence Alignment Library for Next Generation
Sequencing Data
Philipp Rescheneder, Arndt von Haeseler and Fritz J. Sedlazeck
Center for Integrative Bioinformatics Vienna, Max F. Perutz Laboratories, University of Vienna, Vienna, Austria
Medical University of Vienna, Veterinary University of Vienna, Dr. Bohr Gasse 9, 1030 Vienna, Austria
Keywords:
Next generation sequencing, Alignment, High performance computing.
Abstract:
The advent of Next Generation Sequencing (NGS) technologies and the increase in read length and number
of reads per run poses a computational challenge to bioinformatics. The demand for sensible, inexpensive,
and fast methods to align reads to a reference genome is constantly increasing. Due to the high sensitivity
the Smith-Waterman (SW) algorithm is best suited for that. However, its high demand for computational
resources makes it unpractical. Here we present an optimal SW implementation for NGS data and demonstrate
the advantages of using common and inexpensive high performance architectures to improve the computing
time of NGS applications. We implemented a C++ library (MASon) that exploits graphic cards (CUDA,
OpenCL) and CPU vector instructions (SSE, OpenCL) to efficiently handle millions of short local pairwise
sequence alignments (36bp - 1,000bp). All libraries can be easily integrated into existing and upcoming NGS
applications and allow programmers to optimally utilize modern hardware, ranging from desktop computers
to high-end cluster.
1 INTRODUCTION
The advent of Next Generation Sequencing (NGS)
technologies has led to a steep increase in the gener-
ation of sequencing data. Current technologies pro-
duce millions of reads ranging from 36 up to 500
base pairs (Metzker, 2010). Upcoming technologies
such as Ion Torrent and the new Illumina sequencers
will again increase throughput and improve cost ef-
ficiency (Glenn, 2011). In addition, newest Roche
454 and Pacific Biosciences sequencing technologies
will increase the read length to 700 or 1000 base pairs
(Glenn, 2011).
Analyzing NGS data is a demanding task. Keep-
ing pace with the ongoing technological develop-
ments poses a great challenge to bioinformatics. The
first and one of the most limiting steps when an-
alyzing NGS data is mapping reads to a reference
sequence. Current mapping programs like Bowtie
(Langmead et al., 2009) or BWA (Li and Durbin,
2009) are based on the BurrowsWheeler transforma-
tion (BWT). BWT elegantly deals with the sequence
mapping problem. However BWT has an inherent
limitation. The edit distance between read and ref-
erence is limited (Flicek and Birney, 2009). On the
other hand sequence alignments are widely used and
well-studied in the field of genomic research. They
are guaranteed to find an optimal alignment and have
no explicit limitation in terms of edit distance. Be-
cause of this it is desirable to also apply them to
NGS data. Local pairwise alignments (Smith and
Waterman, 1981) are especially suited for this, since
they stop the alignment as soon as too many mis-
matches, insertion or deletions accumulate. However,
their high computational requirements makes them
impractical to use for large data sets. Previous ap-
proaches, e.g. CUDASW++ (Liu et al., 2009), GPU-
Blast (Vouzis and Sahinidis, 2011) or striped Smith-
Waterman (Farrar, 2007)) focused on improving the
performance of one alignment calculation. Contrary
to these methods we focus, here, on a high number
of local alignments to be simultaneously calculated.
This addresses the challenges of processing millions
of reads produced by NGS. Currently over thirty soft-
ware packages are available for processing NGS data
based on pairwise alignment heuristics e.g. SSaha2
(Ning et al., 2001), Shrimp (Rumble et al., 2009)
and BFast (Homer et al., 2009). The general work-
flow is as follows: (1) For each read identify pu-
tative genomic sub regions in the reference genome
195
Rescheneder P., von Haeseler A. and J. Sedlazeck F..
MASon: MILLION ALIGNMENTS IN SECONDS - A Platform Independent Pairwise Sequence Alignment Library for Next Generation Sequencing Data.
DOI: 10.5220/0003775701950201
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2012), pages 195-201
ISBN: 978-989-8425-90-4
Copyright
c
2012 SCITEPRESS (Science and Technology Publications, Lda.)

that show a high similarity to the read. (2) Calculate
alignment scores for all regions found in step 1. (3)
Compute and report the alignment for the best scor-
ing region. Only a few packages, e.g. SARUMAN
(Blom et al., 2011) and MUMmerGPU (Schatz et al.,
2007), exploit the possibilities of modern hardware
like graphic processing units (GPUs). Both require a
Nvidia graphic card. However, none of the over thirty
applications adapt automatically to the hardware that
is available to the user.
Here, we provide MASon, a C++ library us-
ing Nvidia’s Compute Unified Device Architecture
(CUDA) (Kirk and Mei, 2010), the Open Computing
Language (OpenCL) (Stone et al., 2010) and Stream-
ing SIMD Extensions (SSE) (Raman et al., 2000) that
closes the adaptation gap. MASon is an easy to inte-
grate sequence alignment software library that is ca-
pable of handling millions of alignments efficiently,
independent of the underlying hardware. In addition
we show how inexpensive high performance hard-
ware can be used and combined to increase the perfor-
mance of alignment algorithms. Finally we compare
the performanceof the different platforms and discuss
their advantages and disadvantages.
2 METHODS
2.1 Programming Interfaces
To fully utilize every hardware platform ranging from
desktop CPUs and GPUs to highly parallel server pro-
cessors, we are using different application program-
ming interfaces (APIs).
2.1.1 SSE
The Streaming SIMD Extension (SSE) (Raman et al.,
2000) is a single instruction multiple data (SIMD) in-
struction set. SSE was introduced by Intel with the
Pentium III processor. Today, SSE is available on
virtually all computers. The SSE specification con-
sists of several vector instructions that execute a sin-
gle operation on four different floating point variables
in parallel. Originally SSE was designed to speed up
single precision floating point operations as they oc-
cur in image or signal processing. Newer versions of
SSE also support instructions on other data types.
2.1.2 CUDA
The Compute Unified Device Architecture (CUDA)
(Kirk and Mei, 2010) is a C/C++ parallel computing
architecture developed by Nvidia. It enables develop-
ers to write scalable C code in a CPU like manner that
can be executed on a graphic processing unit (GPU).
However, GPUs have a different architecture that is
designed to maximize instruction throughput. This
is achieved by a high number of streaming process-
ing units that are arranged in multiprocessors. Every
graphic card has global memory that is comparable to
CPU memory (RAM). When running computations
on the GPU all data has to reside in global memory (1
to 2 GB). In order to gain performance they can later
be split and moved to more efficient memory types
of the GPU. These memory types are located directly
on each multiprocessor and are orders of magnitudes
faster than global memory. However they are typi-
cally very small (16-48 kb per multiprocessor).
2.1.3 OpenCL
The Open Computing Language (OpenCL) (Stone
et al., 2010) is an open standard that provides a unified
programming interface for heterogeneous platforms
including GPUs and multi core CPUs. In contrast
to CUDA, OpenCL is not limited to a single hard-
ware manufacturer. OpenCL provides an efficient
API that enables software developers to take advan-
tage of the parallel computation capabilities of mod-
ern hardware, such that a single implementation runs
on a wide range of different platforms. The execu-
tion as well as the memory model is very similar to
CUDA.
2.2 Implementation
Computing Smith-Waterman (SW) (Smith and Water-
man, 1981) alignments is very demanding in terms of
runtime and memory resources. The time as well as
the space complexity is O(| R | ∗ | G |). In the case of
NGS R is a read and G is typically a small sub region
of the genome identified by e.g. an exact matching
approach (Ning et al., 2001). The SW algorithm con-
sists of two main steps. First a matrix H is computed.
The maximum of the elements of H is the optimal lo-
cal alignment score for R and G. Starting from this
value, the alignment is reconstructed during the sec-
ond step of the algorithm (backtracking).
For NGS data |R| is small, ranging from 36 to
150 bases for Illumina sequencing. However, usually
there are millions of reads produced by each experi-
ment. When sequencing the human genome with 20-
fold coverageusing Illumina technologies (150bp)the
number of reads m ≈ 400 million. In addition, exact
matching approaches usually report several putative
genomic sub regions per read. For example, when
mapping reads to the human genome the expected
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
196

number n of putative subregions per read is around
n = 180, requiring an exact matching substring of 12.
Thus m ∗ n = 72 billion alignments have to be com-
puted. To deal with this large number one needs to
employ available hardware as efficiently as possible.
For NGS data parallelizing the computation of a sin-
gle alignment is not useful because the computational
cost for a single alignment is low. Therefore we use
a single thread to compute one alignment and focus
on computing as many alignments in parallel as pos-
sible. This brings along the problem that every hard-
ware platform has different capacities when it comes
to parallel computation. On a CPU one can only use
a small number of threads. In contrast a GPU is only
working efficiently when running a high number of
threads in parallel. This means that a minimum num-
ber of alignment computations is required to fully uti-
lize a GPU. However, this minimal number varies be-
tween GPUs. To ensure that our implementation fully
loads different GPUs, independent of their architec-
ture, we calculate the batch size b:
b = N
mp
∗ t
max
(1)
where N
mp
is the number of multiprocessors, t
max
is
the maximum number of threads per multiprocessor.
We furthermore require that
b∗ M
align
≤ M
avail
(2)
where M
align
is the memory required per alignment
and M
avail
is the available global memory on the
graphic card (Nvidia, 2009). If b ∗ M
align
exceeds
M
avail
, b is reduced accordingly. For modern GPUs
b ranges from 50.000 to 100.000 alignment computa-
tions. This high numbers cause the problem that the
memory available for computing a single alignment,
is small. Thus, we implemented a SW alignment that
is less memory demanding. Since most NGS appli-
cation require a reference genome or parts thereof,
e.g. RNA-Seq or they sequence genomes that are
closely related to the reference, we can safely assume
that the number of insertions and deletions between
a read and the reference region is small. Therefore
we implemented a banded alignment algorithm (Gus-
field, 1997). Here only a small corridor surround-
ing the diagonal from upper left to lower right of the
matrix H is computed. Thus the complexity is re-
duced to O(| R | ∗c), where c is the corridor width and
c <<| G |. The memory reduction allows the com-
putation of a high number of alignments such that the
GPU is fully loaded. However, due to its size even the
reduced matrix H has to be stored in global memory.
To further reduce the computing time, we observe
that in NGS mapping only the alignments between
a read and the sub regions of the genome that show
the highest alignment scores are of interest. To iden-
tify the best matching sub region the optimal align-
ment score is sufficient. Thus, the matrix H can be
reduced to a vector of length c and the space com-
plexity is reduced to O(c) (Gusfield, 1997). Note that
the memory usage of score computation is indepen-
dent of read length. However,the computational com-
plexity remains O(r ∗ c) and the corridor width c will
indirectly depend on the read length, since it deter-
mines the number of consecutive insertions and dele-
tions. The reduced memory requirements allow us to
store H in fast on-chip (shared) memory. In addition
all remaining variables, like the scoring function, are
also stored in fast memory (private, constant and tex-
ture). Only read and reference sequences are stored in
global memory. Therefore global memory access is
minimized. This works for all corridor sizes smaller
than 100 on current GPUs. However, since the length
of indels is typically small this restriction of the cor-
ridor size is not limiting the applications.
Although GPUs are powerful co-processing units,
they are not available on every computer. In such
cases a fast computation of alignments on CPUs is de-
sirable. Except for multi-threaded programming, the
most common mechanisms for parallelization on the
CPU is SSE. SSE allows us to speed up score calcula-
tions by processing eight alignmentsin parallel. How-
ever, since SSE only supports a limited set of instruc-
tions, the observed speedup will be lower than eight.
Another major issue is that the backtracking step con-
tains several control flow statements. This makes it
difficult to process more than one alignment concur-
rently. Therefore, the backtracking is done without
SSE.
OpenCL offers another possibility to increase per-
formance on the CPU. As mentioned, OpenCL code
is independent of the hardware. However, to achieve
maximal performance it is necessary to optimize the
SW algorithm for the different platforms (CPU and
GPU). The most important difference between both
platforms is that on the GPU it is important to use
coalescent memory access patterns (Nvidia, 2009),
whereas on the CPU this hinders optimal caching and
therefore degrades performance. The second major
difference is that on the CPU the OpenCL compiler
implicitly uses SSE instructions to increase perfor-
mance. This is only possible when using specific
(vector) data types, which cannot be used efficiently
on a GPU.
In order to allow NGS analysis pipeline program-
mers to employ these technologies, we created a soft-
ware library that consists of three different implemen-
tations. (1) A CPU version including SSE instruc-
tions, (2) a CUDA based version for Nvidia graphic
MASon: MILLION ALIGNMENTS IN SECONDS - A Platform Independent Pairwise Sequence Alignment Library for
Next Generation Sequencing Data
197

Table 1: Runtime (in seconds) of the different library implementations for 1 million local alignment scores. The runtime
required for calculating local alignments is shown in parentheses.
Score computation (Alignment computation)
c = 15 c = 30
Implementations 36 72 100 150 200 500 700 1000
CPU
classical SW -
(9.67)
-
(33.56)
-
(61.42)
-
(132.12)
-
(229.19)
-
(1420)
-
(2737)
-
(5602)
SeqAn -
(6.77)
-
(12.82)
-
(17.49)
-
(25.87)
-
(64.08)
-
(159.59)
-
(223.53)
-
(320.04)
banded SW 3.00
(6.73)
5.95
(12.72)
8.26
(17.33)
12.68
(25.64)
34.61
(63.75)
81.94
(153.88)
114.73
(214.79)
164.09
(305.92)
+ SSE
0.98
(6.73)
1.95
(12.72)
2.71
(17.33)
4.06
(25.64)
10.23
(63.75)
26.37
(153.88)
36.90
(214.79)
52.72
(305.92)
+ OpenCL
1.06
(3.31)
1.97
(6.98)
2.64
(9.38)
3.87
(13.77)
10.33
(36.76)
24.59
(87.28)
34.19
(122.08)
48.90
(171.13)
GPU
+ CUDA
0.07
(0.40)
0.14
(0.77)
0.21
(1.10)
0.33
(1.70)
0.76
(2.79)
1.77
(6.55)
2.47
(9.24)
3.53
(12.68)
+ OpenCL (Nvidia)
0.07
(0.36)
0.14
(0.70)
0.20
(1.01)
0.31
(1.56)
0.67
(2.82)
1.50
(6.76)
2.07
(9.66)
2.96
(14.79)
+ OpenCL (ATI)
0.09
(0.41)
0.16
(0.79)
0.22
(1.08)
0.29
(1.67)
0.79
(3.90)
1.65
(10.52)
2.70
(30.59)
3.24
(886.99)
cards and (3) an OpenCL implementation suitable for
GPUs and CPUs. All of them are encapsulated in
dynamic linked libraries (dlls) also called shared ob-
jects. Programmers can use them by loading the dll
during runtime. This only requires a few lines of code.
Each dll exports functions to calculate the alignment
score and the corresponding alignment. In order to
optimally use the available hardware, the libraries re-
port a recommended batch size (b) for the score and
the alignment calculation.
3 RESULTS & DISCUSSION
To evaluate the runtime of the different implementa-
tions, we created eight simulated datasets with read
lengths ranging from 36bp to 1000bp. Each dataset
consists of 1 million reads and the reference se-
quences. The length of the reference sequences
| G |=| R | +c. The reads contain a 2% sequencing er-
ror consisting of mismatches based on the Jukes Can-
tor model (Jukes and Cantor, 1969), insertions and
deletions. Two computers were used for the runtime
tests. Both are equipped with two 2.6 GHz Intel Xeon
X5650 processors and 32 GB memory. The first com-
puter is equipped with a Nvidia GTX480 card and the
second with a ATI Radeon HD 6970 card. OpenSUSE
is used as operating system.
First we compared the performance of the differ-
ent implementations to the alignment algorithm im-
plemented in the SeqAn (D¨oring et al., 2008) pack-
age. Unfortunately we were not able to get the banded
SW as implemented in SeqAn working. Therefore we
used the semi-global alignment option of the pack-
age as a substitute for comparison. Comparisons are
based on short read (36bp - 150 bp) and long read
(500bp - 1000bp) data. The short read data represents
current and future Illumina and Ion Torrent technolo-
gies, whereas the long read data represents current
and upcoming 454 or Pacific Biosciences technolo-
gies. For aligning short reads we used a corridor size
of 15 otherwise we doubled the corridor width. Table
1 shows the runtimes (in seconds) of computing the
alignment scores and the full alignments for 1 mil-
lion reads. As expected, the banded SW alignment
shows a near linear runtime behavior for a fixed c
whereas the classical SW shows a quadratic runtime
behavior as read length increases. The runtime of the
banded SW alignment and the corresponding SeqAn
algorithm is almost identical. Table 1 also illustrates
the influence of the different programming APIs. For
SSE we observe an average speedup of 3 compared to
the banded SW without SSE. Surprisingly OpenCL
performs slightly better on the CPU than SSE. This
demonstrates that the OpenCL compiler uses SSE in-
struction very efficiently. On a GPU the OpenCL im-
plementation performs slightly better than CUDA for
the score computation. However, the differences are
marginally. Both GPU implementations outperform
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
198

SSE by a factor of 14 to 18.
We are only interested in computing the align-
ment for the genomic region that provides the high-
est score. For these regions backtracking requires ad-
ditional computation time. Comparing the runtimes
of the two banded alignments illustrates the bene-
fits of computing the score without the backtracking
(see table 1). Backtracking slows down the com-
putation by a factor of 2 to 3. OpenCL achieves
a speedup of 1.5 - 2 compared to the banded SW.
Again the graphic card based implementations show
a performance boost (9 − 13 faster) compared to the
CPU (OpenCL) based implementations. In contrast to
the score computation, CUDA outperforms OpenCL
when aligning long reads.
Overall the results demonstrate that OpenCL on
the CPU as well as on the GPU shows the same per-
formance as SSE and CUDA, respectively. Further-
more the runtimes demonstrate a clear advantage of
using the GPU compared to the CPU for aligning
reads and computing scores. However, so far com-
putations are based on a single CPU thread. To in-
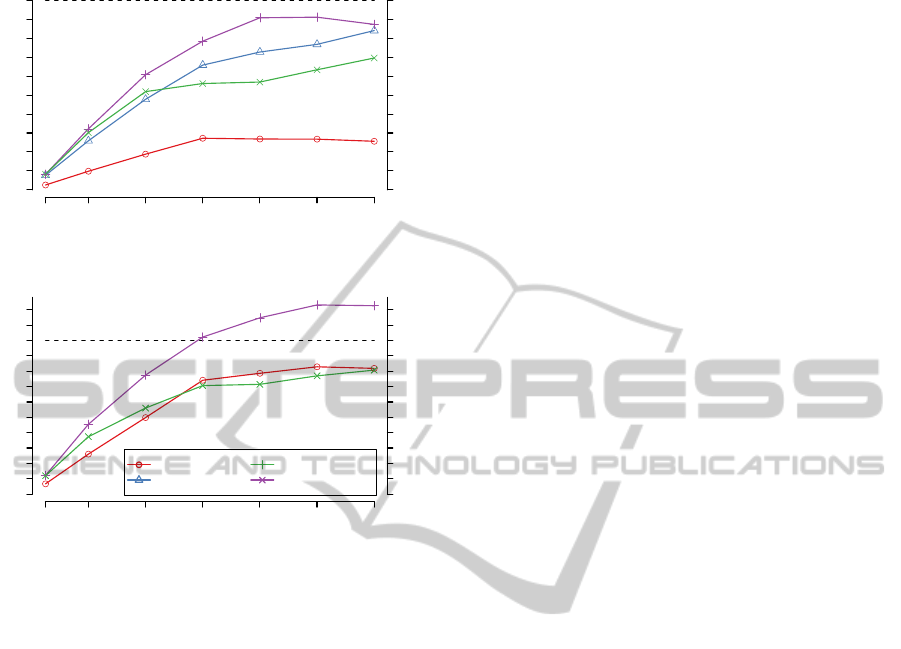
Figure 1: Speedup of the CPU based implementations for an
increasing number of CPU threads. (A) shows the results of
calculating the local alignment scores for 1 million 150bp
sequence pairs.
vestigate how the computation time of our CPU im-
plementations scales with an increasing number of
threads we use OpenMP (Dagum and Menon, 1998)
for parallelization. OpenCL also offers an implicit
parallelization mechanism. Therefore we use two dif-
ferent versions for this comparison. OpenCL CPU1
uses the OpenCL API, whereas OpenCL CPU2 re-
lies on OpenMP to manage the execution on several
CPU cores. The resulting speedups compared to a
single threaded execution for 1 million 150bp reads
are shown in figure 1. Surprisingly the parallelization
using OpenMP performs better than using OpenCL
with multiple cores. Since parallelization is the main
focus of the OpenCL API, this is an unexpected re-
sult. SSE and OpenCL CPU1 perform equally well.
Here we see an almost linear speedup for up to 12
threads. This corresponds to the number of cores
the computer is equipped with (2 x Xeon hexa-core).
Thus it appears that our implementations benefit only
marginally from Intels Hyper-Threading. These re-
sults show that our implementations use the full com-
putational capabilities of a modern multi-core CPU.
In order to compare the computational power
of a CPU to a GPU we measure the number of
scores/alignments computed within the respective
runtime of the fastest GPU implementation. For com-
parison we use an increasing number of threads for
the CPU implementations and take OpenCL (Nvidia)
as baseline (= 100%). Figure 2 shows the results
for 1 million reads of 150bp length. The two Xeon
X5650 hexa-core processors that were used for the
runtime comparison never achieve the performance
of a single GPU (Nvidia GTX 480), when using the
banded algorithm for score computation. When us-
ing the SSE or OpenCL CPU2 implementation the
two processors still compute ∼ 10% less local align-
ment scores than the GPU. For alignment calcula-
tion the OpenCL CPU 2 version outperformsthe GPU
based implementation. However, the two processors
compute only ∼ 20% more alignments than a single
graphic card. Note that a single Xeon X5650 is more
than twice as expensive as a GTX480.
4 CONCLUSIONS
The read length and number of reads cause a compu-
tational problem when analyzing NGS data. There-
fore the demand for fast, flexible and sensitive pro-
cessing software for NGS is obvious. This paper de-
scribes a pairwise local sequence alignment algorithm
(Smith-Waterman) adapted to analyze NGS data. We
show that these optimizations increase the perfor-
mance by a factor of 5 to 57 compared to the banded
SW implementation.
We demonstrate the benefits of using high perfor-
mance programming interfaces (SSE, OpenCL, and
CUDA) when analyzing NGS data. The usage of
such technologies helps us to cope with the increas-
ing throughput of sequencing technologies. By incor-
porating SSE instructions a speedup of 3 to 4 times
is observed without additional costs for processing
units. Furthermore, usage of wide spread and in-
expensive hardware such as graphic cards can sub-
stantially reduce runtime. Using a GPU can improve
the performance of alignment score computation by
a factor of 40 to 55 compared to one CPU thread.
Moreover, our results show that for score computa-
tion one Nvidia GTX480 graphic card outperforms
MASon: MILLION ALIGNMENTS IN SECONDS - A Platform Independent Pairwise Sequence Alignment Library for
Next Generation Sequencing Data
199
(A) Score computation
CPU threads
% scores per 0.3 sec
1 4 8 12 16 20 24
0 20 40 60 80 100
0 20 40 60 80 100
(B) SW alignment
CPU threads
% alignments per 1.6 sec.
1 4 8 12 16 20 24
0 20 40 60 80 100
0 20 40 60 80 100
banded SW
SSE
OpenCL CPU1
OpenCL CPU2
Figure 2: Percentage of score (A) and alignment (B) com-
putations compared to OpenCL (Nvidia) for 1 million 150
bp long sequence pairs.
two Intel Xeon X5650 processors. A computer with
4 graphic cards and 12 CPU cores computes 16 mil-
lion alignment scores or 3.3 million alignments for a
read length of 150 bp in one second. Score computa-
tion for 4∗ 10
8
reads of the length 150 (20-fold cover-
age of the human genome) requires approximately 75
minutes. Thus, even analysis of large NGS data can
be performed in reasonable time.
Since OpenCL allows writing programs for CPUs
as well as GPUs we closer investigated its applica-
bility for sequence alignments. Our results show
that OpenCL outperforms SSE for reads longer than
72bp on the CPU. On the GPU OpenCL outperforms
CUDA for reads smaller than 200bp. Interestingly
OpenCL could not handle multiple CPU threads as ef-
ficiently as OpenMP. However, we are confident that
this will be resolved in the near future.
The key outcome of this work is MASon, a
C++ library that is capable of processing millions
of short (36bp - 1500bp) sequence alignments effi-
ciently. MASon includes SSE and OpenCL based im-
plementations for CPUs as well as GPU based imple-
mentations using CUDA or OpenCL. All implemen-
tations are encapsulated in dynamic linked libraries
and can therefore be easily integrated into existing
or upcoming applications for NGS. By using our li-
brary system every developer can write applications
that optimally utilize modern hardware, ranging from
desktop computers to high-end cluster systems. Fu-
ture work will include extending the libraries with a
global alignment algorithm.
ACKNOWLEDGEMENTS
We would like to thank Hermann Romanek for
help with the implementation and helpful discussion.
Work in A.v.H.’s lab was supported by DFG SPP1174
(grant HA1628-9-2) and the Wiener Wissenschafts
und Technologie Fond (WWTF).
AVAILABILITY
Homepage: http://www.cibiv.at/software/ngm/mason
Operating system(s): Linux, Windows (experimental)
Programming language: C++, CUDA, OpenCL
Other requirements: Graphic card with 1GB memory,
4GB CPU memory
License: Artistic License
REFERENCES
Blom, J., Jakobi, T., Doppmeier, D., Jaenicke, S., Kali-
nowski, J., Stoye, J., and Goesmann, A. (2011). Ex-
act and complete short read alignment to microbial
genomes using GPU programming. Bioinformatics,
27(10):1351–1358.
Dagum, L. and Menon, R. (1998). OpenMP: an industry
standard API for shared-memory programming. IEEE
Computational Science and Engineering, 5(1):46–55.
D¨oring, A., Weese, D., Rausch, T., and Reinert, K. (2008).
SeqAn an efficient, generic C++ library for sequence
analysis. BMC bioinformatics, 9:11.
Farrar, M. (2007). Striped SmithWaterman speeds database
searches six times over other SIMD implementations.
Bioinformatics, 23(2):156–161.
Flicek, P. and Birney, E. (2009). Sense from sequence reads:
methods for alignment and assembly. Nature Meth-
ods, 6(11s):S6–S12.
Glenn, T. C. (2011). Field guide to next-generation DNA se-
quencers. Molecular Ecology Resources, 11(5):759–
769.
Gusfield, D. (1997). Algorithms on strings, trees, and se-
quences : computer science and computational biol-
ogy. Cambridge Univ. Press.
Homer, N., Merriman, B., and Nelson, S. F. (2009).
BFAST: An alignment tool for large scale genome re-
sequencing. PLoS ONE, 4(11):e7767+.
BIOINFORMATICS 2012 - International Conference on Bioinformatics Models, Methods and Algorithms
200

Jukes, T. H. and Cantor, C. R. (1969). Evolution of protein
molecules. Manmmalian Protein Metabolism, pages
21–132.
Kirk, D. B. and Mei, W. (2010). Programming Massively
Parallel Processors: A Hands-on Approach. Morgan
Kaufmann Publishers Inc., San Francisco, CA, USA,
1st edition.
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S.
(2009). Ultrafast and memory-efficient alignment of
short DNA sequences to the human genome. Genome
Biology, 10(3):R25+.
Li, H. and Durbin, R. (2009). Fast and accurate short read
alignment with BurrowsWheeler transform. Bioinfor-
matics, 25(14):1754–1760.
Liu, Y., Maskell, D. L., and Schmidt, B. (2009). CU-
DASW++: optimizing Smith-Waterman sequence
database searches for CUDA-enabled graphics pro-
cessing units. BMC research notes, 2(1):73+.
Metzker, M. L. (2010). Sequencing technologies - the next
generation. Nature reviews. Genetics, 11(1):31–46.
Ning, Z., Cox, A. J., and Mullikin, J. C. (2001). SSAHA: a
fast search method for large DNA databases. Genome
research, 11(10):1725–1729.
Nvidia (2009). NVIDIA CUDA C Programming Best Prac-
tices Guide. Nvidia, 1st edition.
Raman, S. K., Pentkovski, V., and Keshava, J. (2000). Im-
plementing streaming SIMD extensions on the pen-
tium III processor. IEEE Micro, 20(4):47–57.
Rumble, S. M., Lacroute, P., Dalca, A. V., Fiume, M.,
Sidow, A., and Brudno, M. (2009). SHRiMP: Accu-
rate mapping of short color-space reads. PLoS Comput
Biol, 5(5):e1000386+.
Schatz, M., Trapnell, C., Delcher, A., and Varshney, A.
(2007). High-throughput sequence alignment us-
ing graphics processing units. BMC Bioinformatics,
8(1):474+.
Smith, T. F. and Waterman, M. S. (1981). Identification of
common molecular subsequences. Journal of molec-
ular biology, 147(1):195–197.
Stone, J. E., Gohara, D., and Shi, G. (2010). OpenCL:
A parallel programming standard for heterogeneous
computing systems. Computing in Science & Engi-
neering, 12(3):66–73.
Vouzis, P. D. and Sahinidis, N. V. (2011). GPU-BLAST: us-
ing graphics processors to accelerate protein sequence
alignment. Bioinformatics, 27(2):182–188.
MASon: MILLION ALIGNMENTS IN SECONDS - A Platform Independent Pairwise Sequence Alignment Library for
Next Generation Sequencing Data
201
