
Learning Advanced TFBS Models from Chip-Seq Data
diChIPMunk: Effective Construction of Dinucleotide Positional Weight Matrices
Ivan V. Kulakovskiy
1,2
, Victor G. Levitsky
3,4
, Dmitry G. Oschepkov
3
, Ilya E. Vorontsov
2,5
and Vsevolod J. Makeev
2,6,7
1
Laboratory of Bioinformatics and Systems Biology, Engelhardt Institute of Molecular Biology,
Russian Academy of Sciences, Vavilov str. 32, Moscow, 119991, GSP-1, Russia
2
Department of Computational Systems Biology, Vavilov Institute of General Genetics, Russian Academy of Sciences,
Gubkina str. 3, Moscow, 119991, Russia
3
Laboratory of Molecular Genetics Systems, Institute of Cytology and Genetics of the Siberian Division of Russian
Academy of Sciences, Lavrentiev Prospect 6, Novosibirsk, 630090, Russia
4
Faculty of Natural Sciences, Novosibirsk State University, Pirogova str. 2, Novosibirsk, 630090, Russia
5
Yandex Data Analysis School, Data Analysis Department, Moscow Institute of Physics and Technology,
Leo Tolstoy Str. 16, Moscow, 119021, Russia
6
State Research Institute of Genetics and Selection of Industrial Microorganisms,
1st Dorozhny proezd, 1 Moscow, 117545, Russia
7
Moscow Institute of Physics and Technology, Institutskii per. 9, Dolgoprudny, 141700, Moscow Region, Russia
Keywords: Motif Discovery, Transcription Factor Binding Sites, TFBS Models, Positional Weight Matrices, PWM,
ChIP-Seq, Dinucleotide Composition.
Abstract: Identification and consequent analysis of DNA sequence motifs recognized by transcription factors is an
important component in studying transcriptional regulation in higher eukaryotes. In particular, motif
discovery methods are applied to construct transcription factor binding sites (TFBSs) models. The TFBS
models are then used for prediction of putative binding sites in genomic regions of interest. The most
popular TFBS model is a positional weight matrix (PWM). The PWM is usually constructed from
nucleotide positional frequencies estimated from a gapless multiple local alignments of experimentally
identified TFBS sequences. Modern high-throughput experiments, like ChIP-Seq, provide enough data for
careful training of more advanced models having more parameters. Until now, the majority of existing tools
for TFBS prediction in ChIP-Seq data still rely on PWMs with independent positions. This is partly
explained with only marginal improvement of specificity and sensitivity of TFBS recognition for advanced
models over those based on traditional PWMs if trained on ChIP-Seq data. Here we present a novel
computational tool, diChIPMunk (http://autosome.ru/dichipmunk/), which can construct dinucleotide
PWMs accounting for neighboring nucleotide correlations in input sequences. diChIPMunk retains
advantages of the published ChIPMunk algorithm, including usage of ChIP-Seq peak shape and overall
computational efficiency. Using public ChIP-Seq data for several TFs we show that carefully trained
dinucleotide PWMs perform significantly better as compared to PWMs based on mononucleotide
frequencies.
1 INTRODUCTION
Our understanding of transcription regulation
mechanisms in higher eukaryotes is far from
complete. One of the most studied mechanisms is
driven by transcription factors (TFs) recognizing
specific sites at DNA. Modern high-throughput
methods allow detecting tens of thousands of DNA
segments that are bound by particular protein in
particular conditions. With the wet-lab supplying an
immense amount of data special computational tools
are required to detect text patterns (also called as
DNA motifs) that correspond to binding sites (BSs)
of TFs under consideration. The current stage of
experimental technologies requires motif discovery
in silico for accurate identification of TF binding
pattern from any type of experimental data. The
146
V. Kulakovskiy I., G. Levitsky V., G. Oschepkov D., E. Vorontsov I. and J. Makeev V..
Learning Advanced TFBS Models from Chip-Seq Data - diChIPMunk: Effective Construction of Dinucleotide Positional Weight Matrices.
DOI: 10.5220/0004238201460150
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2013), pages 146-150
ISBN: 978-989-8565-35-8
Copyright
c
2013 SCITEPRESS (Science and Technology Publications, Lda.)

TFBS models produced during this step can be
consequently applied for computational prediction of
TFBSs in genomic regions of interest. The most
popular model, a positional weight matrix (PWM), is
inferred directly from a gapless local multiple
alignment of sequences of TF-bound DNA regions
(Stormo, 2000). The elements of the matrix (the
positional weights) at individual motif positions are
assumed independent. Till now many methods to
detect DNA motifs in ChIP-Seq data were published
(Thomas-Chollier et al., 2012, Suppl. table 1) but
most of them were based on simple mononucleotide
PWMs. Recent attempts to use ChIP-Seq data to
construct more complex models (e.g. TPD, Bi et al.,
2011) resulted in TFBS recognition quality that was
not significantly better comparing to simple PWMs
with independent positional weights.
There is a specific family of TFBS models with
non-independent positional weights that take into
account correlations of nucleotides occupying
neighboring positions within TFBSs. This
correlation agrees well with the role of neighboring
nucleotides in formation of DNA structure
(SantaLucia and Hicks, 2004). PWM based on
dinucleotide statistics is the most straightforward of
models that take into account interaction of
neighboring nucleotides. Previously it has been
shown that the dinucleotide PWMs perform
significantly better than classic mononucleotide
PWMs if a training set of sequences is large enough
(Gershenzon et al., 2005) and (Levitsky et al., 2007).
Moreover, experiments with protein-binding
microarrays were successfully explained by
producing TFBS models that take into account
frequencies of neighboring dinucleotides (Zhao et
al., 2012). So it appears fruitful to try a similar
approach for analysis of ChIP-Seq data, which also
provides enough information to gather a sufficiently
large training set.
As a starting point we adopted ChIPMunk
algorithm as a state of the art tool that performed
well in our own (Kulakovskiy et al., 2010) and
several independent benchmark studies (Ma et al.,
2012) and (Kuttippurathu et al., 2011). The
advantage of ChIPMunk is that it takes into account
ChIP-Seq base coverage data (the shape of reads
pileup that points to probable locations of binding
sites under ChIP-Seq peaks). In this study we
present a novel tool, diChIPMunk, which produces a
dinucleotide PWM (diPWM defining a Markov
order 1 model of TFBS motif) that incorporates
information on dependencies between nucleotides in
neighboring alignment positions. We show how the
dinucleotide PWMs can be included into the
ChIPMunk algorithm framework. We also show
results of tests demonstrating that usage of
dinucleotide PWMs significantly improves TFBS
recognition quality in ChIP-Seq data.
2 METHODS
diChIPMunk algorithm is constructed on top of a
subsampling-based greedy optimization procedure.
A random starting diPWM and a corresponding
gapless multiple local alignment are optimized on a
random subset of the initial sequence set (taking the
best diPWM hits from each sequence). The obtained
diPWM is then reoptimized on the full sequence set.
Greedy PWM optimization converges rather
quickly, the described two-step optimization
procedure allows further improvement of
convergence speed and offers a simple solution for
the classic problem of getting stuck at a local
optimum. Thus the algorithm core is almost the
same as in ChIPMunk (Kulakovskiy et al., 2010).
Neighboring positions in the diPWM are not
independent since each single nucleotide is included
in two overlapping dinucleotides. diChIPMunk
converts all sequences from mono- to dinucleotide
alphabet of 16 letters where each letter represents a
dinucleotide (AA, AC, AG, .., TT). For example,
AACC sequence is written as A-A-C-C in
nucleotides and AA-AC-CC in dinucleotides. The
tricky point here is that all sequences over ACGT-
letter alphabet constitute only a subset of all
sequence over AA-to-TT-alphabet since the second
nucleotide of the first dinucleotide must be the same
as the first nucleotide of the second dinucleotide and
so on. I.e. dinucleotide sequence AC-CG
unambiguously maps to nucleotide sequence A-C-G,
but there is no ACGT-alphabet counterpart for
dinucleotide sequence AC-AG.
2.1 KDIDIC
To search for an optimal diPWM diChIPMunk tests
each putative gapless multiple local alignment if it
contains highly conservative dinucleotide columns
(i.e. with the dinucleotide distribution far from
uniform having some highly prevalent
dinucleotides). In ChIPMunk the Kullback Discrete
Information Content was used to make a criterion
for the alignment optimality. With the sequences
written in dinucleotide alphabet a similar measure
can be used to estimate alignment quality for
dinucleotides (Kullback Dinucleotide Discrete
Information Content, KDIDIC):
LearningAdvancedTFBSModelsfromChip-SeqData-diChIPMunk:EffectiveConstructionofDinucleotidePositional
WeightMatrices
147

KDIDIC
,
!N!
∈
AA,..TT
,
∈AA,..TT
(1)
Here
is the letter in dinucleotide alphabet; q
is the
background frequency of
; j is the position within
gapless local multiple alignment; x
,j
is the
frequency of dinucleotide
in j-th column of the
alignment; l is the length (the width) of the
alignment and N is the total number of aligned
sequences. The alignment with the maximal
KDIDIC value is considered optimal.
This measure has a maximum for some
alignment over all possible sequences written in 16
letter dinucleotide alphabet. We are interested in a
subset of sequences that can be mapped to sequences
written in 4-letter ACGT-letter alphabet.
Equation (1) is used by diChIPMunk to provide an
easy-to-compute estimation of the deviation of
dinucleotide frequencies in a given alignment from a
given background dinucleotide distribution q
.
KDIDIC-optimal dinucleotide model from
diChIPMunk should perform stably better than
mononucleotide PWM. This is confirmed by our
tests presented in the Results section below.
2.2 Estimating Alignment Width
To estimate the optimal length of the aligned
segments of TFBS sequences, the alignment width,
and the corresponding diPWM length diChIPMunk
uses an heuristic procedure that locates the longest
strong motif in a given lengths range. The motif is
called strong if the first and the last columns of the
corresponding alignment have KDIDIC no less than
a predefined threshold. The threshold value was
arbitrary selected as equal to KDIDIC calculated for
a column missing 2 arbitrary dinucleotide letters and
having frequencies of all 14 remaining dinucleotides
uniformly distributed. The procedure yielded motif
lengths comparable to those of mononucleotide
ChIPMunk (see examples in Figure 1).
2.3 Benchmarking Datasets
We used ChIP-Seq data from ENCODE: data for
AP2A, GATA1 TFs (Yale ChIP-Seq, base coverage
profile available) and REST, GABPA TFs
(HudsonAlpha ChIP-Seq, no base coverage data).
The datasets were taken from the HOCOMOCO
database (Kulakovskiy et al., 2012). For each dataset
the subset of top 1000 peaks was taken and sorted
according to peak height value. 500 peaks with even
ranks were used for motif discovery. 500 peaks with
odd ranks were used as an independent positive
control set consisting of sequences not involved in
construction of TFBS models. The datasets used for
TFBS model construction and independent positive
control datasets are available on the diChIPMunk
website.
2.4 Benchmarking Procedure
For each TF we compared three models. The longest
PWM from TRANSFAC (Matys et al., 2006)
database was used as a baseline for comparison. If
several models with the same width were presented
in TRANSFAC we selected the one constructed
from the largest set of binding sites. Two other
TFBS models were PWM obtained by the
ChIPMunk algorithm and diPWM obtained by
diChIPMunk algorithm. Local nucleotide
(dinucleotide) composition was used by ChIPMunk
(diChIPMunk) as a background model for motif
discovery on ChIP-Seq without base coverage data
(REST and GABP TFs). Motif lengths range was set
as 10 to 25bps. The overall benchmarking procedure
was similar to that presented in (Kulakovskiy et al.,
2012).
True Positive (TP) rate was estimated from the
number of sequences from the independent control
set having PWM hits scoring no less than the
threshold. For a full spectrum of TP rates for each
TFBS model we then estimated a set of
corresponding score thresholds. For each threshold
we computed P-value which represented the fraction
of all DNA segments that are recognized as binding
sites by the model (see Section 2.5). P-value can be
interpreted as the probability to obtain a score no
less than the threshold in a particular position of a
random DNA sequence. To estimate P-values for
mono- and diPWMs we have used an approach from
(Touzet and Varre, 2007) reimplemented in
MACRO-APE (http://autosome.ru/dimacroape/).
Thus we can estimate the False Positive (FP) rate
as the probability to find at least one PWM hit with a
score no less than the threshold in a random double-
strand DNA segment of a fixed length L:
FP 1‐
1‐
P
‐value
(2)
Here L is selected as the median sequence length
estimated from the independent control set, l is the
PWM length and the PWM hits are assumed to be
independent with their total number complying
compound Poisson distribution.
Having a set of TP rates and FP estimates for
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
148

each model we plotted a ROC curve for each TF and
computed an area-under-curve (AUC) value that
allows comparing TFBS recognition quality.
3 RESULTS AND CONCLUSIONS
Figure 1 shows ROC curves comparing diPWMs
versus mononucleotide PWMs constructed from the
same ChIP-Seq data and existing TRANSFAC
PWMs. Motif LOGO representations are given.
AUC values are presented directly on graphs.
diPWMs clearly outperformed models based on
single nucleotide PWMs for all tested datasets (see
Figure 1). However, previously it was shown that
not all TFs profit from diPWMs (Levitsky, 2007) so
a more comprehensive study of various ChIP-Seq
datasets remains highly important.
We have estimated computational performance
of diChIPMunk versus its mononucleotide precursor
using 4 threads for Core i7 CPU. Since a
dinucleotide model has more parameters to train the
default number of starting random seeds and
subsampling runs is doubled for diChIPMunk. The
computationsl performance was acceptable (1 to 8
hours to train the diPWM including length
estimation; the absolute values for mononucleotide
models of ChIPMunk are 2 to 4 times better).
Dinucleotide models derived from ChIP-Seq data
performed significantly better than their
mononucleotide analogs in four independent ChIP-
Seq datasets. Dinucleotide models require more
computational power to be carefully trained, but it is
still possible even using a desktop computer. With
the increasing availability of different types of high-
throughput data we suspect the improved models
becoming widely used. The next step is open for
novel post-processing tools that would allow model
comparison and effective genome-scale prediction of
binding sites.
ACKNOWLEDGEMENTS
This work was supported by a Dynasty Foundation
Fellowship [to I.V.K.]; Russian Foundation for
Basic Research [12-04-32082 to I.V.K.] and [12-04-
01736-a to D.O.]; Presidium of the Russian
Academy of Sciences program in Cellular and
Molecular Biology.
REFERENCES
Stormo, G. D., (2000). DNA binding sites: representation
and discovery. Bioinformatics, 16(1):16-23.
Thomas-Chollier, M., Darbo, E., Herrmann, C., Defrance,
M., Thieffry, D., van Helden, J., (2012). A complete
workflow for the analysis of full-size ChIP-seq (and
similar) data sets using peak-motifs. Nat Protoc.,
7(8):1551-68.
Bi, Y., Kim, H., Gupta, R., Davuluri, R. V., (2011). Tree-
based position weight matrix approach to model
transcription factor binding site profiles. PLoS One.,
6(9):e24210.
SantaLucia., J. Jr., Hicks, D., (2004). The thermodynamics
of DNA structural motifs. Annu Rev Biophys Biomol
Struct., 33:415-40.
Gershenzon, N. I., Stormo, G. D., Ioshikhes, I. P., (2005).
Computational technique for improvement of the
position-weight matrices for the DNA/protein binding
sites. Nucleic Acids Res., 33(7):2290-301.
Levitsky, V. G., Ignatieva, E. V., Ananko, E. A., Turnaev,
I. I., Merkulova, T. I., Kolchanov, N. A., Hodgman, T.
C. (2007). Effective transcription factor binding site
prediction using a combination of optimization, a
genetic algorithm and discriminant analysis to capture
distant interactions. BMC Bioinformatics, 8:481.
Zhao, Y., Ruan, S., Pandey, M., Stormo, G. D. (2012).
Improved models for transcription factor binding site
identification using nonindependent interactions.
Genetics, 191(3):781-90.
Kulakovskiy, I. V., Boeva, V. A., Favorov, A. V.,
Makeev, V. J. (2010). Deep and wide digging for
binding motifs in ChIP-Seq data. Bioinformatics,
26(20):2622-3.
Kulakovskiy I. V., Medvedeva Y. A., Schaefer U.,
Kasianov A. S., Vorontsov I. E., Bajic V.B., Makeev
V. K., (2012) HOCOMOCO: a comprehensive
collection of human transcription factor binding sites
models. Nucleic Acids Res, in press.
Ma, X., Kulkarni, A., Zhang, Z., Xuan, Z., Serfling, R.,
Zhang, M. Q. (2012). A highly efficient and effective
motif discovery method for ChIP-seq/ChIP-chip data
using positional information. Nucleic Acids Res.,
40(7):e50.
Kuttippurathu, L., Hsing, M., Liu, Y., Schmidt, B.,
Maskell, D. L., Lee, K., He, A., Pu, W. T., Kong, S.
W., (2011). CompleteMOTIFs: DNA motif discovery
platform for transcription factor binding experiments.
Bioinformatics, 27(5):715-7.
Matys, V., Kel-Margoulis, O. V., Fricke, E., Liebich, I.,
Land, S., Barre-Dirrie, A., Reuter, I., Chekmenev, D.,
Krull, M., Hornischer, K., Voss, N., Stegmaier, P.,
Lewicki-Potapov, B., Saxel, H., Kel, A. E.,
Wingender, E., (2006). TRANSFAC and its module
TRANSCompel: transcriptional gene regulation in
eukaryotes. Nucleic Acids Res., 34(Database
issue):D108-10.
Touzet, H., Varré, J. S. (2007). Efficient and accurate P-
value computation for Position Weight Matrices.
Algorithms Mol Biol., 11;2:15.
LearningAdvancedTFBSModelsfromChip-SeqData-diChIPMunk:EffectiveConstructionofDinucleotidePositional
WeightMatrices
149
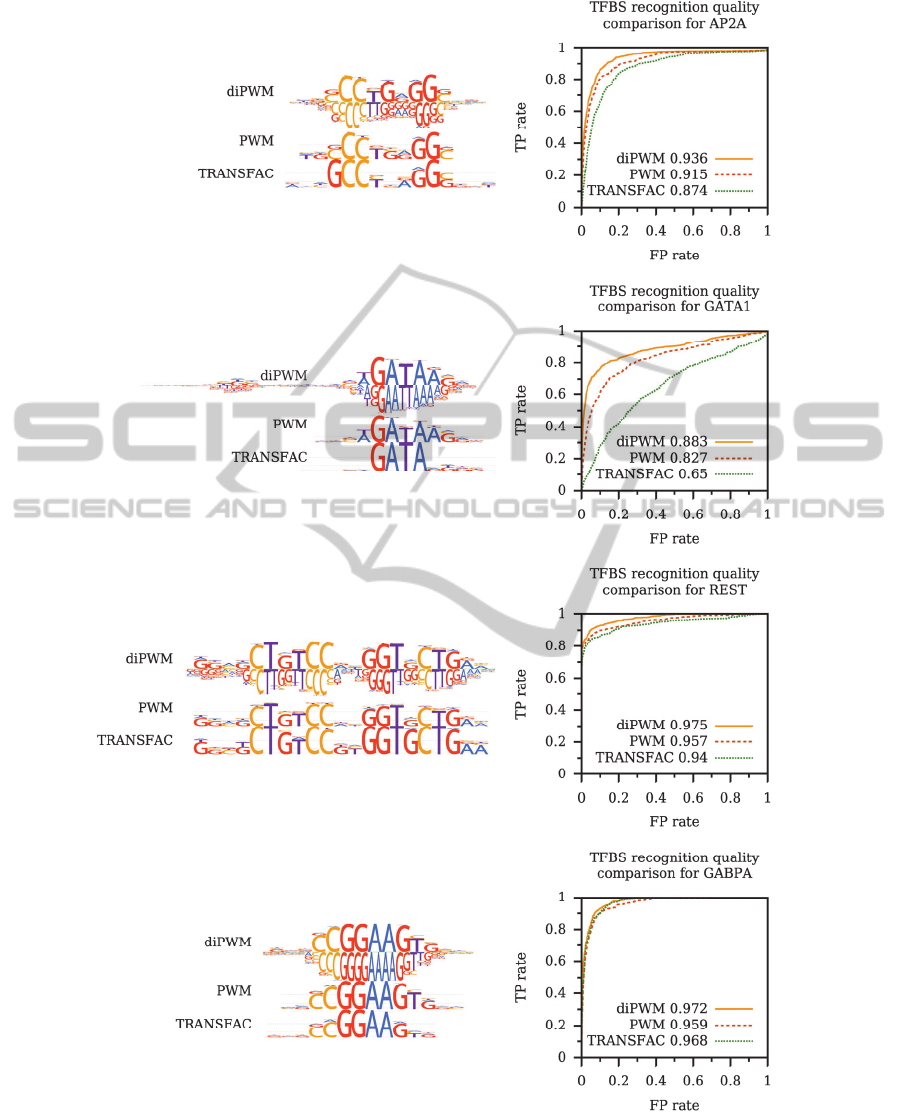
Figure 1: Comparison of TFBS recognition quality for mono- and dinucleotide PWMs obtained from ChIP-Seq data;
TRANSFAC PWM as the baseline model. ROC curves displaying True Positive rate (TP rate) versus False Positive rate
(FP rate) are shown on the right panels with the corresponding AUC (area-under-curve) values. Motif LOGO
representations are given on the left panels. Higher curves (and higher AUC values) correspond to models with better
recognition quality (i.e. higher TP rate for a fixed FP rate). It is notable, that diChIPMunk versus ChIPMunk comparison
shows AUC improvement comparable to that of ChIPMunk versus TRANSFAC.
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
150
