
De Novo Short Read Assembly Algorithm with Low Memory Usage
Yuki Endo
1
, Fubito Toyama
1
, Chikafumi Chiba
2
, Hiroshi Mori
1
and Kenji Shoji
1
1
Graduate School of Engineering, Utsunomiya University, Yoto 7-1-2, Utsunomiya, Tochigi, 321-8585, Japan
2
Faculty of Life and Environmental Sciences, University of Tsukuba, Tennoudai 1-1-1, Tsukuba, Ibaraki 305-8572, Japan
Keywords:
Bioinfomatics, Next Generation Sequencing, De Novo Assembly
Abstract:
Determining whole genome sequences of various species has many applications not only in biological system,
but also in medicine, pharmacy and agriculture. In recent years, the emergence of high-throughput next-
generation sequencing technologies has dramatically reduced time and costs for whole genome sequencing.
These new technologies provide ultrahigh throughput with lower unit data cost. However, the data are very
short length fragments of DNA. Thus, developing algorithms for merging these fragments is very important.
Merging these fragments without reference data is called de novo assembly. Many algorithms for de novo
assembly have been proposed in recent years. Velvet, one of the algorithms, is famous because it has good
performance in terms of memory and time consumption. But memory consumption increases dramatically
when the size of input fragments is huge. Therefore, it is necessary to develop algorithm with low memory
usage. In this paper, we propose an algorithm for de novo assembly with lower memory. In our experiments
using E.coli K-12 strain MG 1655, memory consumption of the proposed algorithm was one-third of that of
Velvet.
1 INTRODUCTION
Determining whole genome sequences of various
species has many applications not only in biologi-
cal system, but also in medicine, pharmacy and agri-
culture. In recent years, the emergence of high-
throughput next-generation sequencer (NGS) tech-
nologies has dramatically reduced time and costs for
whole genome sequencing. These new technologies
provide ultrahigh throughput with lower unit data
cost. However, many very small fragments (fewer
than 100 base pair) of DNA are sequenced in these
technologies. The fragments are typically called
reads. The precision of NGS is not perfect. Therefore,
the reads obtained by NGS might include sequencing
errors. Thus, developingalgorithms for mergingthese
reads is very important. Merging these reads without
reference data is called de novo assembly.
The de novo assembly algorithms can be classi-
fied into two approaches by its features : overlap-
layout-consensus (OLC) and de Bruijn graph. OLC
approaches relies on an overlap graph. Edena (Her-
nandez et al., 2008), MIRA (Chevreux et al., 2004),
Celera (Miller et al., 2008), SSAKE (Warren et al.,
2007), and VCAKE (Jeck et al., 2007) assemblers
are based on OLC approach. In OLC strategy, pair-
wise overlaps are found by all-againtst-all pair-wise
comparison. Overlap graphs are constructed from
pair-wise overlaps. In overlap graphs, a vertex rep-
resents a read and an edge denotes an overlap be-
tween two connected vertices (reads). Consensus se-
quences are determined by tracing paths in the graph.
On the other hand, Velvet (Zerbino and Birney, 2008),
ABySS (Simpson et al., 2009), ALLPATHS (Butler
et al., 2008), and SOAPdenovo (Li et al., 2010) as-
semblers are based on de Bruijn graph approach. In
the de Bruijn graph, a vertex represents a sequence of
k bases (k-mer), where k is any positive integer. An
edge represents an overlap of k-1 bases. Thus, two
connected vertices denote a k-1 overlap between their
vertices (k-mers). After the de Bruijn graph was con-
structed from reads obtained by NGS, contigs are de-
termined by tracing paths in the graph. The de Bruijn
graph approach is most widely applied to the short
reads from Solexa and SOLiD platforms. In this ap-
proach, fixed-length (k-1) overlaps are found and re-
dundant k-mers (subsequence) are compressed. Thus,
it is suitable for assembling vast quantities of short
reads. Velvet is one of the most popular de novo as-
sembler based on the de Bruijn graph. However mem-
ory consumption increases dramatically when the size
of input reads is huge (more than several gigabytes).
Therefore it is hard to use Velvet for huge scale as-
sembly.
215
Endo Y., Toyama F., Chiba C., Mori H. and Shoji K..
De Novo Short Read Assembly Algorithm with Low Memory Usage.
DOI: 10.5220/0004881002150220
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2014), pages 215-220
ISBN: 978-989-758-012-3
Copyright
c
2014 SCITEPRESS (Science and Technology Publications, Lda.)

In this paper, we propose an algorithm for huge
scale de novo assembly with low memory usage. Al-
though our algorithm is based on de Bruijn graph ap-
proach in the same way as Velvet, edge information
is not kept in main memory. Thus, the amount of
memory can be reduced greatly in our method. In
our experiments using E.coli K-12 strain MG 1655,
the results showed that maximum memory consump-
tion of the proposed algorithm was one-third that of
Velvet. Furthermore, the running time of proposed
method was also faster than that of Velvet.
2 AN ASSEMBLY ALGORITHM
WITH LOW MEMORY USAGE
In this paper, we propose an algorithm for huge scale
de novo assembly with low memory usage. Figure 1
shows the outline of our algorithm. First, all k-mers
obtained by dividing reads are recorded. At the same
time, the number of occurrences of each k-mer is also
counted. Second, the de Bruijn graph is constructed
using k-mers. Then, the graph is partitioned into sub-
graphs such that the subgraph has a simple path or a
simple cycle. The simple path is a path in the graph
which does not have repeating vertices or edges. The
subgraphs are connected to make a larger simple path.
The number of occurrences of a k-mer is used to se-
lect a connection path. Finally, contigs are generated
by tracing vertices in the each connected graphs.
2.1 Extraction of k-mers
k-mers are extracted from all reads. They are kept in
database in memory as “k-mer integer”. As shown
in Table 1, k-mer integer is a representation of one-
to-one correspondence between k-mer and integer.
Specifically, bases “A”, “C”, “G”, and “T” correspond
to quaternary digits 0, 1, 2, and 3 respectively. Thus, a
k-mer sequence is expressed as a quaternary numeral.
For example, a 5-mer base sequence “ACGTA” is con-
verted to the quaternary number 01230. Then, it is
converted to the decimal number 108. Thus, 108 is
k-mer integer corresponding to “ACGTA”. If a k-mer
sequence is represented by a string, memory would
be needed k bytes. In the k-mer integer representa-
tion, the amount of memory for k-mer sequences can
be reduced to one fourth of that of the string repre-
sentation. Using k-mer integer is not only superior
to memory usage, but also to processing time. For-
ward and reversecomplement k-mer sequences are re-
garded as the same sequence in our method. Either of
the two complementary sequences is registered in the
database on main memory.
Figure 1: Outline of the proposed method.
In this work, k-mer integer and the number of oc-
currences of the k-mer corresponding to the k-mer in-
teger are kept in main memory. In order to achieve
low memory usage, other data (such as edges in de
Bruijn graph) is not kept in the memory. K-mer se-
quence in which the number of occurrences of the
sequence is small (less than a threshold) is not used
in the graph construction because it is suspected that
such k-mer sequences contain sequencing errors. In
our experiments, the threshold was set to 5. Figure
2 shows the extraction of k-mer and the contents of
database in the proposed method.
BIOINFORMATICS2014-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
216
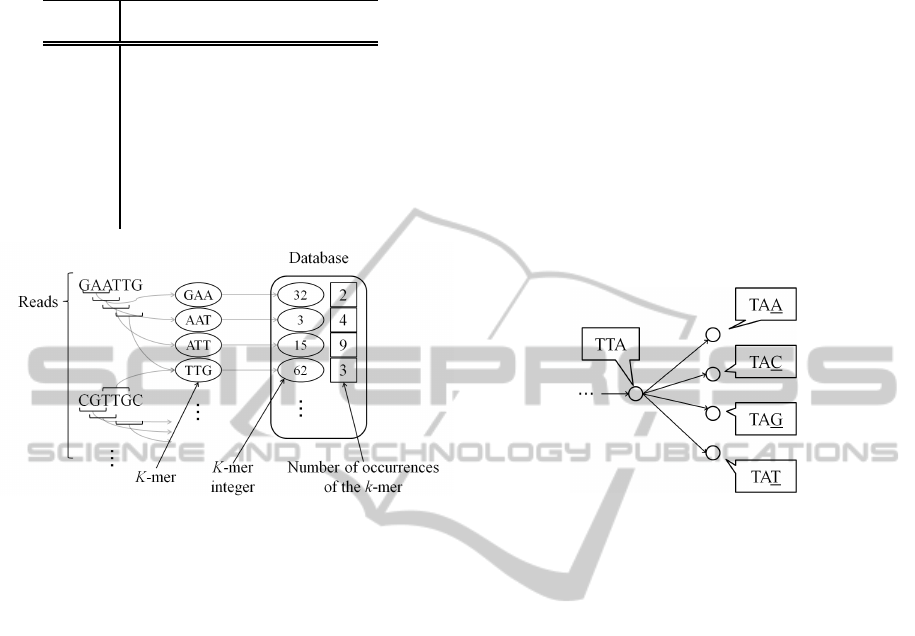
Table 1: k-mer sequence corresponding to k-mer integer
(in case of 5-mer).
k-mer Quaternary Binary
k-mer integer
(decimal)
AAAAA 0 0 0
AAAAC
1 1 1
AAAAG
2 10 2
AAAAT
3 11 3
AAACA
10 100 4
AAACC
11 101 5
AAACG
12 110 6
AAACT
13 111 7
AAAGA
21 1000 8
AAAGC
22 1001 9
Figure 2: Extraction of k-mer and the contents of database
in the proposed method (in case of 3-mer).
2.2 Graph Construction
The de Bruijn graph is constructed using k-mers. In
the de Bruijn graph, each vertex represents a k-mer.
An edge represents an overlap of k-1 bases. Thus,
two connected vertices denote a k-1 overlap between
their vertices (k-mers). For example, there is an edge
between the two vertices corresponding to “ACGTA”
and “CGTAC”. The direction of the edge is from
“ACGTA” to “CGTAC”.
The contigs are generated by tracing vertices in
the de Bruijn. However, de Bruijn graph constructed
from short reads has numerous branches and cycles.
Therefore, it is difficult to find the paths from which
contigs are constructed. The details of the method to
find out such paths are described in Section 2.3.
In conventional algorithms using de Bruijn graph,
when the graph is constructed, edge information
about which vertices are connected to each other is
also kept in main memory. Since there are many
edges in the graph, keeping the edge information con-
sumes a huge amount of memory. In this paper,
the edge information is not kept in main memory.
Thus, although computational time for assembly is
increased, memory usage can be reduced. The ex-
istence of the edge is calculated only when it is re-
quired. Specifically, the vertices which are connected
by a directed edge from a vertex have only 4 types of
k-mers because the k-mers which are represented by
the connected vertices overlap by k-1 bases as shown
in Fig. 3. Thus, the connected vertices (k-mer se-
quences) can be obtained by checking four values of
k-mer integer in the database. By using this method,
although processing time to check the values of k-mer
integer is increased, memory for keeping connection
edge data is not required. Only the data of vertices
are kept in the database. Thus, the amount of mem-
ory can be reduced greatly in our method. The graph
construction is finished by registering the values of k-
mer integer from k-mer sequences and the number of
occurrences of each k-mer on our database.
Figure 3: Example of 4 types of k-mers which are connected
to the current vertex (in case of 3-mer).
2.3 Edge Removal
As mentioned in Section 2.2, the constructed graph
has numerous branches and cycles. Consequently,
it is important to select the edges on the path from
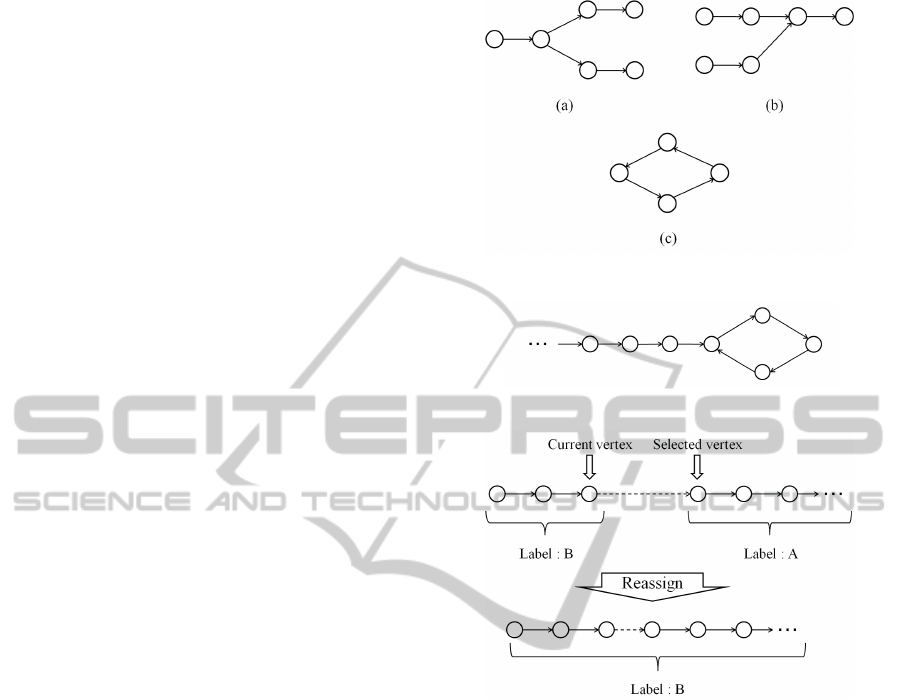
which a contig is constructed. Figure 4 shows exam-
ples of branches. A vertex has multiple edges in Fig.
4 (a). A vertex has multiple edges from other vertices
in Fig. 4 (b). Although the outdegree of the vertices
with branches is two in Fig. 4, the maximum number
of the out degree is four. Figure 4 (c) shows an ex-
ample of a cycle. Actually, these branches and cycles
are intricately intertwined. A path from the directed
graph in which branches and cycles are included is
needed to generate a contig. It is necessary to deter-
mine the unique simple path based on some criteria.
The edge removal process is as follows.
1. A start vertex (k-mer) which has the largest num-
ber of occurrences is selected.
2. The start vertex is set to the current vertex.
3. The existence of the vertices which are connected
to the current vertex is checked.
(a) If one connected vertex is found, the vertex is
set to the current vertex. Go to 3.
(b) If multiple connected vertices are found, one of
them is set to the current vertex. The details of
DeNovoShortReadAssemblyAlgorithmwithLowMemoryUsage
217
the vertex selection are described in later in this
section. Go to 3.
(c) If the connected vertex is not found, the current
vertex is regarded as the end vertex. Go to 4.
4. The existence of the vertices which are not se-
lected yet is checked.
(a) If the vertices which are not selected are found,
the new start vertex which has the largest num-
ber of occurrences is selected from the vertices
which are not selected yet. Go to 2.
(b) If the vertices which are not selected are not
found, the process is finished.
In this process, the vertices which are included in a
path are assigned to same label. A path from the start
vertex to the end vertex represents a subgraph. Multi-
ple subgraphs are created in this process;
The details of the vertex selection in which
branches and cycles are included are described as be-
low. When there are multiple out going edges from
the current vertex as shown Fig. 4 (a), the edge con-
nected to the vertex in which the number of occur-
rences of k-mer is the largest is selected and the other
outgoing edges are removed. When there are multi-
ple incoming edges as shown in Fig. 4 (b), the edge
is selected by the same process and other incoming
edges are removed. In this edge selection process,
when the difference of occurrences of k-mer between
the selected vertex and unselected vertices is less than
a threshold, the current vertex is regarded as the end
vertex and new start vertex is selected again. When
the label of the selected vertex is the same as that of
the current vertex as shown in Fig. 4 (c) (in the case
of cycle), the current vertex is regarded as the end ver-
tex. In many cases, a cycle is a part of a path as shown
in Fig. 5. The vertices which are in the cycle are as-
signed to the new label in this case. Thus, a cycle is
separated from the current scanning path.
Furthermore, there is the case in which the se-
lected vertex has been assigned to other label as
shown in Fig. 6. In this case, all the vertices in the
path which has the selected vertex are reassigned to
the label of the current vertex.
2.4 Subgraph Connection and Contig
Construction
To construct a longer path, subgraphs obtained by the
process described in previous section are connected.
The outline of the subgraph connection process is as
follows. First, a subgraph with simple path is se-
lected. The subgraphs with the longest path is se-
lected from subgraphs which have not been selected.
Figure 4: Examples of branches and cycle.
Figure 5: Example of a cycle which is a part of a path.
Figure 6: Example of the label reassignment.
However the subgraph with a simple cycle can be se-
lected more than once. This subgraph is set to the start
subgraph. Next, the subgraph in which the start vertex
or the end vertexare connected to the end vertexor the
start vertex of the selected subgraph are searched. The
details of connection of the subgraph with a simple
cycle are described in later in this section. This pro-
cess is realized by checking k-mer integer described
in the previous section. If such subgraph is found,
the start (end) vertex is connected to the end (start)
vertex, and the two subgraph are merged into a sin-
gle subgraph. This graph expanding process is re-
peated while merging graph exists. If there are multi-
ple subgraphs which can be connected, the subgraph
with longer simple path is connected. Figure 7 shows
an example of connecting subgraphs. The example
in this figure is the case of left side connection. The
same process is repeated on the right side.
In addition, the subgraph with a simple cycle is
connected as shown in Fig. 8. In this figure, The ver-
tex v
a
is start vertex of subgraph G
path
with a simple
path. The vertex v
b
is contained in subgraph G
cycle
BIOINFORMATICS2014-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
218

with a simple cycle. If the k-mers of the vertex v
a
and
v
b
overlap each other, the vertex v
c
which connected
to v
b
is checked. (v
c
is also contained G
cycle
.) v
b
and
v
c
are regarded as the start( or end) vertex. Then, the
G
path
and G
cycle
are merged. If the vertex v
c
is con-
nected to other vertex which is included in other sub-
graph, These subgraphs are merged again as shown in
Fig. 8.
Figure 7: Example of subgraphs connection.
Figure 8: Example of the connection of subgraph with a
simple cycle.
After the subgraph connection, the list of the ver-
tices is obtained by tracing the path which is included
in a subgraph. The contig is generated by merging k-
mers which are referenced from the vertices as shown
in Fig. 9. The final contigs are obtained by repeating
this process for all subgraphs. In our experiments, the
contigs which are longer than a threshold are output.
In our experiments, the threshold was set to length of
the reads.
3 EXPERIMENTAL RESULTS
To evaluate the performance of the proposed method,
we compare the performance of our method with that
of Velvet which is one of the most popular de novo
assembler based on the de bruijn graph. E.coli K-
12 strain MG 1655 for which the complete DNA se-
quence is known was used in our experiments. The
Figure 9: Generation of contigs.
sequence length is approximately 4.6Mbp. The real
dataset with 35bp reads was made by a computer from
the complete DNA sequence of E.coli K-12 strain
MG 1655. Experiments were conducted using reads
(35bp) that generated by the NGS from the genome.
Experiments were run with the k-mer size of 19, 21,
23, 25, 27, 29, and 31. We assessed the maximum
memory consumption, the running time, the contig
length, and the accuracy of contigs.
Figure 10 and Fig. 11 show the maximum mem-
ory usage and the running time for each k-mer, re-
spectively. As shown in Fig. 10, the memory usage of
the proposed algorithm is one-third of that of Velvet.
Therefore, the purpose of this research was achieved
because the amount of memory was reduced greatly.
Moreover the average running time of the proposed
method was approximately 41% of that of Velvet. In
the proposed method, the connection between vertices
is checked each time when a path is traced. Thus,
the running time to find the connection between ver-
tices is increased. However, since the path tracing
algorithms for resolving branches and cycles is very
simple, the average running time was faster in the
proposed method. In addition, the running time was
shortened according to the increase of the k-mer size.
On the other hand, the change of the k-mer size had
hardly influence on the memory.
Table 2 shows the results of the contig quantity of
the proposed method and Velvet. Maximum length,
N50 length, the number of contigs, and total bases
are shown in Table 2. The N50 length is defined as
the length of the shortest contig such that the sum of
contigs of equal length or longer is at least 50% of
the total length of all contigs. Table 3 shows the re-
sults of the accuracy of the contigs. Percentage of
the genome covered and average error rate are shown
in Table 3. The maximum length of the proposed
method was shorter than that of Velvet as shown in Ta-
ble 2. Moreover, N50 length of the proposed method
was shorter than that of Velvet. The main reason for
these results is that the path tracing algorithm for re-
DeNovoShortReadAssemblyAlgorithmwithLowMemoryUsage
219

solving branches and cycles is very simple in the pro-
posed method. Thus, the maximum length and N50
length would be better by modifying the path trac-
ing algorithm. On the other hand, the error rate of
the proposed method was lower than that of Velvet.
The genome coverage was slightly lower than that of
Velvet. These results indicate that there are not large
differences in the contig quantity from path tracing
algorithms.
Figure 10: Comparison of maximum memory consumption.
Figure 11: Comparison of running time.
Table 2: Comparison of quantity of contigs.
Maximum
length (bp)
N50
(bp)
# of
contigs
Total
(bp)
Proposed method 74,708 17,038 631 4,560,202
Velvet
81,421 22,870 754 4,544,229
Table 3: Comparison of precision of contigs.
Genome
covered (%)
Average
error rate (%)
Proposed method 99.59 5.71
Velvet
99.89 7.29
4 CONCLUSIONS
In this paper, we propose an algorithm for huge scale
de novo assembly with low memory usage. In our ex-
periments using E.coli K-12 strain MG 1655, the re-
sults showed that maximum memory consumption of
the proposed algorithm was one-third that of Velvet.
Furthermore, the running time of proposed method
was also faster than that of Velvet. These results
showed that the proposed method outperformed Vel-
vet for the memory and the running time. On the other
hand, contig quality obtained by the proposed method
was slightly worse than that of Velvet. To improve
the accuracy of the contigs, we need to modifying the
path tracing algorithm in the future works.
REFERENCES
Butler, J., MacCallum, I., Kleber, M., Shlyakhter, I. A.,
Belmonte, M. K., Lander, E. S., Nusbaum, C., and
Jaffe, D. B. (2008). ALLPATHS: de novo assembly
of whole-genome shotgun microreads. Genome Res.,
18(5):810–820.
Chevreux, B., Pfisterer, T., Drescher, B., Driesel, A. J.,
Muller, W. E., Wetter, T., and Suhai, S. (2004). Us-
ing the miraEST assembler for reliable and automated
mRNA transcript assembly and SNP detection in se-
quenced ESTs. Genome Res., 14(6):1147–1159.
Hernandez, D., Francois, P., Farinelli, L., Osteras, M., and
Schrenzel, J. (2008). De novo bacterial genome se-
quencing: millions of very short reads assembled on a
desktop computer. Genome Res., 18(5):802–809.
Jeck, W. R., Reinhardt, J. A., Baltrus, D. A., Hicken-
botham, M. T., Magrini, V., Mardis, E. R., Dangl,
J. L., and Jones, C. D. (2007). Extending assembly
of short DNA sequences to handle error. Bioinformat-
ics, 23(21):2942–2944.
Li, R., Zhu, H., Ruan, J., Qian, W., Fang, X., Shi, Z., Li,
Y., Li, S., Shan, G., Kristiansen, K., Li, S., Yang, H.,
Wang, J., and Wang, J. (2010). De novo assembly
of human genomes with massively parallel short read
sequencing. Genome Res., 20(2):265–272.
Miller, J. R., Delcher, A. L., Koren, S., Venter, E., Walenz,
B. P., Brownley, A., Johnson, J., Li, K., Mobarry,
C., and Sutton, G. (2008). Aggressive assembly of
pyrosequencing reads with mates. Bioinformatics,
24(24):2818–2824.
Simpson, J. T., Wong, K., Jackman, S. D., Schein, J. E.,
Jones, S. J., and Birol, I. (2009). ABySS: a parallel
assembler for short read sequence data. Genome Res.,
19(6):1117–1123.
Warren, R. L., Sutton, G. G., Jones, S. J., and Holt, R. A.
(2007). Assembling millions of short DNA sequences
using SSAKE. Bioinformatics, 23(4):500–501.
Zerbino, D. R. and Birney, E. (2008). Velvet: algorithms for
de novo short read assembly using de Bruijn graphs.
Genome Res., 18(5):821–829.
BIOINFORMATICS2014-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
220
